2 附錄XIX A 藥品質量標準分析方法驗證指導原則
藥品質量標準分析方法驗證的目的是證明採用的方法適合於相應檢測要求。在建立藥品質量標準時,分析方法需經驗證;在藥品生產工藝變更、製劑的組分變更、原分析方法進行修訂時,則質量標準分析方法也需進行驗證。方法驗證理由、過程和結果均應記載在藥品質量標準起草說明或修訂說明中。
需驗證的分析項目有:鑑別試驗、雜質定量檢查或限度檢查、原料藥或製劑中有效成分含量測定,以及製劑中其他成分(如防腐劑等)的測定。藥品溶出度、釋放度等檢查中,其溶出量等的測試方法也應做必要驗證。
驗證內容有:準確度、精密度(包括重複性、中間精密度和重現性)、專屬性、檢測限、定量限、線性、範圍和耐用性。視具體方法擬訂驗證的內容。附表中列出的分析項目和相應的驗證內容可供參考。
2.1 一、準確度
準確度係指用該方法測定的結果與真實值或參考值接近的程度,一般用回收率(%)表示。準確度應在規定的範圍內測試。
原料藥可用已知純度的對照品或供試品進行測定,或用本法所得結果與已知準確度的另一個方法測定的結果進行比較。
製劑可用含已知量被測物的各組分混合物進行測定。如不能得到製劑的全部組分,可向製劑中加入已知量的被測物進行測定,或用本法所得結果與已知準確度的另一個方法測定結果進行比較。
如該分析方法已經測試並求出了精密度、線性和專屬性,在準確度也可推算出來的情況下,這一項可不必再做。
2.雜質定量測定的準確度
可向原料藥或製劑中加入已知量雜質進行測定。如不能得到雜質或降解產物,可用本法測定結果與另一成熟的方法進行比較,如藥典標準方法或經過驗證的方法。在不能測得雜質或降解產物的響應因子或不能測得對原料藥的相對響應因子的情況下,可用原料藥的響應因子。應明確表明單個雜質和雜質總量相當於主成分的重量比(%)或面積比(%)。
3.數據要求
在規定範圍內,至少用9個測定結果行評價。例如,設計3個不同濃度,每個濃度各分別製備3供試品溶液,進行測定。應報告已知加入量的回收率(%),或測定結果平均值與真實值之差及其相對標準偏差或可信限。
2.2 二、精密度
精密度係指在規定的測試條件下,同一個均勻供試品,經多次取樣測定所得結果之間的接近程度。精密度一般用偏差、標準偏差或相對標準偏差表示。
在相同條件下,由同一個分析人員測定所得結果的精密度稱爲重複性;在同一個實驗室,不同時間由不同分析人員用不同設備測定結果之間的精密度,稱爲中間精密度;在不同實驗室由不同分析人員測定結果之間的精密度,稱爲重現性。
1.重複性
在規定範圍內,至少用9個測定結果進行評價。例如,設計3個不同濃度,每個濃度各分別製備3份供試品溶液,進行測定,或將相當於100%濃度水平的供試品溶液,用至少測定6次的結果進行評價。
2.中間精密度
爲考察隨機變動因素對精密度的影響,應設計方案進行中間精密度試驗。變動因素爲不同日期、不同分析人員、不同設備。
3.重現性
法定標準採用的分析方法,應進行重現性試驗。例如,建立藥典分析方法時,通過協同檢驗得出重現性結果。協同檢驗的目的、過程和重現性結果均應記載在起草說明中。應注意重現性試驗用的樣品本身的質量均勻性和貯存運輸中的環境影響因素,以免影響重現性結果。
4.數據要求
均應報告標準偏差、相對標準偏差和可信限。
2.3 三、專屬性
專屬性係指在其他成分(如雜質、降解產物、輔料等)可能存在下,採用的方法能正確測定出被測物的特性。鑑別反應、雜質檢查和含量測定方法,均應考察其專屬性。如方法不夠專屬,應採用多個方法予以補充。
1.鑑別反應
應能與可能共存的物質或結構相似化合物區分。不含被測成分的供試品,以及結構相似或組分中的有關化合物,應均呈負反應。
2.含量測定和雜質測定
色譜法和其他分離方法,應附代表性圖譜,以說明方法的專屬性,並應標明諸成分在圖中的位置,色譜法中的分離度應符合要求。
在雜質可獲得的情況下,對於含量測定,試樣中可加入雜質或輔料,考察測定結果是否受干擾,並可與未加雜質或輔料的試樣比較測定結果。對於雜質測定,也可向試樣中加入一定量的雜質,考察雜質之間能否得到分離。
在雜質或降解產物不能獲得的情況下,可將含有雜質或降解產物的試樣進行測定,與另一個經驗證了的方法或藥典方法比較結果。用強光照射、高溫、高溼、酸(鹼)水解或氧化的方法進行加速破壞,以研究可能的降解產物和降解途徑。含量測定方法應比對二法的結果,雜質檢查應比對檢出的雜質個數,必要時可採用光二極管陣列檢測和質譜檢測,進行峯純度檢查。
2.4 四、檢測限
檢測限係指試樣中被測物能被檢測出的最低量。藥品的鑑別試驗和雜質檢查方法,均應通過測試確定方法的檢測限。常用的方法如下。
1.非儀器分析目視法
用已知濃度的被測物,試驗出能被可靠地檢測出的最低濃度或量。
2.信噪比法
用於能顯示基線噪聲的分析方法,即把已知低濃度試樣測出的信號與空白樣品測出的信號進行比較,算出能被可靠地檢測出的最低濃度或量。一般以信噪比爲3:1或2:1時相應濃度或注入儀器的量確定檢測限。
3.數據要求
應附測試圖譜,說明測試過程和檢測限結果。
2.5 五、定量限
定量限係指試樣中被測物能被定量測定的最低量,其測定結果應具一定準確度和精密度。雜質和降解產物用定量測定方法研究時,應確定方法的定量限。
常用信噪比法確定定量限。一般以信噪比爲10:1時相應濃度或注入儀器的量確定定量限。
2.6 六、線性
線性係指在設計的範圍內,測試結果與試樣中被測物濃度直接呈正比關係的程度。
應在規定的範圍內測定線性關係。可用一貯備液經精密稀釋,或分別精密稱樣,製備一系列供試樣品的方法進行測定,至少製備5份供試樣品。以測得的響應信號作爲被測物濃度的函數作圖,觀察是否呈線性,再用最小二乘法進行線性迴歸。必要時,響應信號可經數學轉換,再進行線性迴歸計算。
2.7 七、範圍
範圍係指能達到一定精密度、準確度和線性,測試方法適用的高低限濃度或量的區間。
範圍應根據分析方法的具體應用和線性、準確度、精密度結果和要求確定。原料藥和製劑含量測定,範圍應爲測試濃度的80%~120%;製劑含量均勻度檢查,範圍應爲測試濃度的70%~130%,根據劑型特點,如氣霧劑和噴霧劑,範圍可適當放寬;溶出度或釋放度中的溶出量測定,範圍應爲限度的±20%,如規定了限度範圍,則應爲下限的-20%至上限的+20%;雜質測定,範圍應根據初步實測,擬訂爲規定限度的±20%。如果含量測定與雜質檢查同時進行,用百分歸一化法,則線性範圍應爲雜質規定限度的-20%至含量限度(或上限)的+20%。
2.8 八、耐用性
耐用性係指在測定條件有小的變動時,測定結果不受影響的承受程度,爲使方法可用於常規檢驗提供依據。開始研究分析方法時,就應考慮其耐用性。如果測試條件要求苛刻,則應在方法中寫明。典型的變動因素有:被測溶液的穩定性、樣品的提取次數、時間等。液相色譜法中典型的變動因素有:流動相的組成和pH值、不同廠牌或不同批號的同類型色譜柱、柱溫、流速等。氣相色譜法變動因素有:不同廠牌或批號的色譜柱、固定相、不同類型的擔體、柱溫、進樣口和檢測器溫度等。
經試驗,應說明小的變動能否通過設計的系統適用性試驗,以確保方法有效。
3 附錄XIX B 藥物製劑人體生物利用度和生物等效性試驗指導原則
生物利用度是指製劑中的藥物被吸收進入血液的速率和程度。生物等效性是指一種藥物的不同製劑在相同的試驗條件下,給以相同的劑量,反映其吸收速率和程度的主要動力學參數沒有明顯的統計學差異。
口服或其他非脈管內給藥的製劑,其活性成分的吸收受多種因素的影響。包括製劑工藝、藥物粒徑、晶型或多晶型,處方中的賦形劑、黏合劑、崩解劑、潤滑劑、包衣材料、溶劑、助懸劑等。生物利用度是保證藥品內在質量的重要指標,而生物等效性則是保證含同一藥物的不同製劑質量一致性的主要依據。生物利用度與生物等效性概念雖不完全相同,但試驗方法基本一致。爲了控制藥品質量,保證藥品的有效性和安全性,特制定本指導原則。何種藥物製劑需要進行生物等效性或生物利用度試驗,可根據有關部門頒佈的法規要求進行。
進行藥物製劑人體生物利用度和生物等效性試驗的臨牀實驗室和分析實驗室,應提供機構名稱以及醫學、科學或分析負責人的姓名、職稱和簡歷。
3.1 一、生物樣品分析方法的基本要求
生物樣品中藥物及其代謝產物定量分析方法的專屬性和靈敏度,是生物利用度和生物等效性試驗成功的關鍵。首選色譜法,如HPLC、GC以及GC-MS、LC-MS、LC-MS-MS聯用技術,一般應採用內標法定量。必要時也可採用生物學方法或生物化學方法。
由於生物樣品取樣量少、藥物濃度低、內源性物質(如無機鹽、脂質、蛋白質、代謝物)及個體差異等多種因素影響生物樣品測定,所以必鬚根據待測物的結構、生物介質和預期的濃度範圍,建立適宜的生物樣品分析方法,並對方法進行驗證。
1.專屬性
必須證明所測定的物質是原形藥物或特定的活性代謝物,內源性物質和相應的代謝物不得干擾樣品的測定。對於色譜法至少要提供空白生物樣品色譜圖、空白生物樣品外加對照物質色譜圖(註明濃度)及用藥後的生物樣品色譜圖。對於複方製劑應特別加強專屬性研究,以排除可能的干擾。對於LC-MS和LC-MS-MS方法,應着重考察基質效應。
2.標準曲線與線性範圍
根據所測定物質的濃度與響應的相關性,用迴歸分析方法獲得標準曲線。標準曲線高低濃度範圍爲線性範圍,在線性範圍內濃度測定結果應達到試驗要求的精密度和準確度。必須用至少6個濃度建立標準曲線,應使用與待測樣品相同的生物介質,線性範圍要能覆蓋全部待測濃度,不允許將線性範圍外推求算未知樣品的濃度。標準曲線不包括零點。
要求選擇3個濃度的質控樣品同時進行方法的精密度和準確度考察。低濃度接近定量下限(LLOQ),在LLOQ的3倍以內;高濃度接近於標準曲線的上限;中間選一個濃度。每一濃度至少測定5個樣品。
精密度用質控樣品的日內和日間相對標準差(RSD)表示,RSD一般應小於15%,在LLOQ附近RSD應小於20%。準確度是指用特定方法測得的生物樣品濃度與真實濃度的接近程度,一般應在85%~115%範圍內,在LLOQ附近應在80%~120%範圍內。
4.定量下限
定量下限是標準曲線上的最低濃度點,要求至少能滿足
測定3~5個半衰期時樣品中的藥物濃度,或Cmax的1/10~1/20時的藥物濃度,其準確度應在真實濃度的80%~120%範圍內,RSD應小於20%,信噪比應大於5。
5.樣品穩定性
根據具體情況,對含藥生物樣品在室溫、冰凍和凍融條件下以及不同存放時間進行穩定性考察,以確定生物樣品的存放條件和時間。
6.提取回收率
應考察高、中、低3個濃度的提取回收率,其結果應一致、精密和可重現。
7.質控樣品
質控樣品系將已知量的待測藥物加入到生物介質中配製的樣品,用於質量控制。
8.質量控制
應在生物樣品分析方法驗證完成之後開始測試未知樣品。每個未知樣品一般測定一次,必要時可進行復測。生物樣品每個分析批測定時應建立新的標準曲線,並隨行測定高、中、低3個濃度的質控樣品,每個濃度多重樣本。每個分析批質控樣品數不得少於未知樣品數的5%,且不得少於6個。質控樣品測定結果的偏差一般應小於15%,低濃度點偏差一般應小於20%。最多允許33%的質控樣品結果超限,且不得均在同一濃度。如不合格,則該分析批樣品測試結果作廢。
9.測試結果
應詳細描述所用的分析方法,引用已有的參考文獻,提供每天的標準曲線、質控樣品及未知樣品的結果計算過程。還應提供全部未知樣品分析的色譜圖,包括全部相關的標準曲線和質控樣品的色譜圖,以供審查。
3.2 二、普通製劑
3.2.1 (一)受試者的選擇
對參加生物利用度和生物等效性研究的受試者有一些特殊的要求。
1.受試者條件
一般情況選健康男性,特殊情況說明原因,如婦科用藥。兒童用藥應在成人中進行。具體要求如下。
(1)性別爲男性。
(2)年齡一般要求18~40歲;同一批試驗受試者年齡不宜相差10歲或以上。
(3)體重與標準體重相差±10%,同一批試驗受試者體重應相近,體重單位以千克(kg)計。
(4)身體健康,無心、肝、腎、消化道、神經系統疾病及代謝異常等病史,並進行健康體檢(如檢查心電圖、血壓、心率、肝功能、腎功能、肺功能和血常規等),應無異常。特殊藥物還需要檢查相應的其他指標,如降血糖藥物應檢查血糖水平。
(6)2周前至試驗期間不服用其他任何藥物,試驗期間禁菸、酒及含咖啡因的飲料。
(7)試驗單位應與志願受試者簽署知情同意書。
2.受試者的例數
受試者必須具有足夠的例數,按有關規定要求18~24例,必要時可增加受試者人數。
3.2.2 (二)參比製劑
生物利用度和生物等效性研究,必須有參比製劑作對照。參比製劑的安全性和有效性應該合格,參比製劑選擇的原則如下:進行絕對生物利用度研究時選用上市的靜脈注射劑爲參比製劑;進行相對生物利用度或生物等效性研究時,應選擇國內外同類上市主導產品作爲參比製劑。
3.2.3 (三)受試製劑
受試製劑應爲符合臨牀應用質量標準的放大試驗產品,應提供受試製劑和參比製劑的體外溶出度比較(n≥12)數據,以及穩定性、含量或效價等數據。個別藥物尚需提供多晶型及光學異構體資料。
受試和參比製劑實測含量差異應在5%之內。
3.2.4 (四)試驗設計
對於兩個製劑,即一個爲受試製劑,另一個爲參比製劑,通常採用雙週期兩製劑交叉試驗設計,以抵消試驗週期和個體差異對試驗結果的影響。即將受試者隨機分成兩組,一組先服用受試製劑,後服用參比製劑;另一組先服用參比製劑,後服用受試製劑。兩個試驗週期之間爲洗淨期,洗淨期應不少於藥物的10個半衰期,通常爲1周或2周。
對於3個製劑,即兩個受試製劑和一個參比製劑,此時宜採用3製劑、3週期的二重3×3拉丁方式試驗設計。每個週期之間的洗淨期通常爲1周或2周。
取樣點對試驗的可靠性起着重要作用。服藥前取空白血樣。一個完整的血藥濃度一時間曲線應包括吸收相、分佈相和消除相,總採樣(不包括空白)不少於12個點。取樣一般持續到3~5個半衰期或血藥濃度爲cmax的1/10~1/20。
在不能用血藥濃度測定時,可採用其他生物樣品進行測定,如尿液,但試驗藥品與試驗方案應符合生物利用度測定要求。
3.2.5 (五)服藥劑量確定
進行生物利用度與生物等效性研究時,藥物劑量一般應與臨牀用藥劑量一致。受試製劑和參比製劑最好應用等劑量。如需使用不相等劑量時,應說明原因,若藥物在此劑量範圍內符合線性動力學規律,則計算生物利用度時應以劑量校正。
3.2.6 (六)研究過程
受試者禁食過夜(10小時以上),於次日早晨空腹服用受試製劑或參比製劑,用250ml溫開水送服。服藥2小時後方可飲水,4小時後進統一標準餐。受試者於服藥後,按要求在不同時間取靜脈血。根據需要取血樣(血漿、血清或全血),並冷凍貯存,備測。受試者服藥後避免劇烈活動。取血樣在臨牀監護室中進行。如受試者有不良反應時應有應急措施,必要時應停止試驗。
生物等效性首選在禁食狀態下進行,但對於空腹給藥生物利用度非常低或者易出現胃腸道功能紊亂等強烈副作用的藥物,可改爲餐後給藥進行生物等效性試驗。
3.2.7 (七)藥物動力學分析
將所得的各受試者不同時間樣品的血藥濃度數據及平均值與標準差列表並作圖,然後分別對各受試者進行有關藥物動力學參數求算,並求出參數的平均值和標準差。主要的藥物動力學參數爲消除半衰期(t1/2)、峯濃度(cmax)、峯時間(tmax)和血藥濃度一時間曲線下面積AUC。cmax、tmax用實測值表示,不得內推。AUC0→tn(零到t時間的血藥濃度-時間曲線下面積)用梯形法或對數梯形法計算,tn是最後一次可測濃度的取樣時間。AUC0→∞(零到無限大時間的血藥濃度-時間曲線下面積)按下式計算,AUC0→∞=AUC0→tn+Ctn/λz。 ctn是最後一點的血藥濃度,λz是末端消除速度常數。λz用對數血藥濃度-時間曲線線末端直線部分的斜率求得,t1/2=0.693/λz求出。

3.2.8 (八)生物利用度計算
1.單次給藥
生物利用度F應用各個受試者的AUC0→tn和AUC0→∞分別計算,並求其均值與標準差。受試製劑(T)和參比製劑(R)劑量相同時:
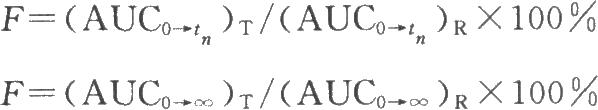
若受試藥物具有線性藥物動力學特徵時,受試製劑和參比製劑也可採用不同劑量,並按下式予以校正。
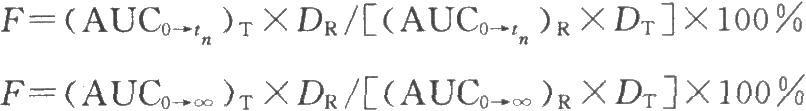
式中 DR爲參比製劑的給藥劑量;
DT爲受試製劑的給藥劑量。
代謝產物數據:對於前體藥物,或由於藥物在體內代謝極快,無法測定血中原形藥物,此時可採用相應的活性代謝物進行生物利用度研究。
生物利用度計算以AUC0→tn爲主,參考AUC0→∞
2.多次給藥
在下列情況下,可考慮多次給藥達穩態後,用穩態血藥濃度估算生物利用度:(1)藥物吸收程度相差不大,但吸收速度有較大差異;(2)生物利用度個體差異大;(3)緩釋、控釋製劑;(4)當單次給藥後原藥或代謝產物濃度很低,不能用相應的分析方法準確測得。
多次給藥,經等間隔(τ)給藥至穩態後,在某一給藥間隔時間內,多次採集樣品,分析藥物濃度,計算在穩態劑量間隔期間從0~τ時間的血藥濃度一時間曲線下的面積(AUCSS)。當受試製劑和參比製劑劑量相等時,即可用下式求得相對生物利用度。

式中,AUCSST和AUCSSR分別代表受試製劑與參比製劑穩態條件下的AUC。
3.2.9 (九)生物等效性評價(藥物動力學數據統計分析)
應對藥物動力學主要參數(如AUC、cmax)進行統計分析,作出生物等效性評價。統計分析先將AUC和cmax數據進行對數轉換,然後進行方差分析與雙單側t檢驗處理,若受試製劑和參比製劑AUC幾何均值比的90%置信區間在80%~125%範圍內,且cmax幾何均值比的90%置信區間在75%~133%範圍內,則判定受試製劑與參比製劑生物等效。tmax可用非參數法進行檢驗。
爲了便於生物等效性比較,每一受試者服用不同製劑的藥物動力學參數(AUC和cmax)應平行列表,還要列出受試製劑(T)和參比製劑(R)參數之間的比值(T/R)和比值的對數。除了計算它們的算術均值外,還應該計算幾何均值,都應該包括在報告中。
3.2.10 (十)臨牀報告、副作用和不良反應
受試者病史、身體檢查和化驗結果,以及與研究相關的可能副作用和不良反應,均應報告。
3.3 三、緩釋、控釋製劑
緩釋、控釋製劑的生物利用度與生物等效性試驗應在單次給藥與多次給藥兩種條件下進行。進行該類製劑生物等效性試驗的前提是應進行至少3種溶出介質的兩者體外溶出行爲同等性研究。
3.3.1 (一)單次給藥雙週期交叉試驗
本試驗目的是在受試者空腹條件下,比較受試製劑與參比製劑的吸收速率和吸收程度,確認受試緩釋、控釋製劑與參比製劑是否爲生物等效,並具有緩釋、控釋特徵。
1.受試者要求與選擇標準同普通製劑。
2.參比製劑
一般應選用國內外上市的同類緩釋、控釋製劑主導產品爲參比製劑。若系創新的緩釋、控釋製劑,則應選擇國內外上市的同類普通製劑主導產品爲參比製劑。
3.試驗過程
同普通製劑單次給藥。
4.提供數據
(1)列出各受試者的血藥濃度一時間數據、血藥濃度平均值與標準差,列表並作圖。
(2)計算各受試者藥物動力學參數並求平均值與標準差。cmax、tmax、AUC0→tn、AUC0→tn和F;儘可能提供其他參數如平均滯留時間(MRT)等。
5.生物等效性評價
若受試緩釋、控釋製劑與參比緩釋、控釋製劑比較,AUC、Cmax符合生物等效性要求,tmax統計上應無顯著差異,則認爲兩種製劑生物等效。若受試緩釋、控釋製劑與普通製劑比較,AUC符合生物等效性要求(同普通製劑AUC生物等效性評價),則認爲吸收程度生物等效;若cmax有所降低,tmax有所延長,並按“二、普通製劑(九)”進行統計分析,其結果至少有一項指標不符合生物等效時,則表明受試製劑有緩釋或控移特徵。
3.3.2 (二)多次給藥雙週期交叉試驗
本試驗目的是研究受試緩釋、控釋製劑與參比製劑多次給藥達穩態的速率與程度以及穩態血藥濃度的波動情況。
1.受試者要求及選擇標準
同單劑量項下,可繼續用單劑量的受試者。受試者至少爲18~24例,必要時可以適當增加。參比製劑同單次給藥。
2.試驗設計及過程
採用隨機交叉試驗設計方法,多次服用受試製劑與參比製劑。對於受試製劑,用擬定的用藥劑量和方案。每日1次用藥的製劑,受試者應在空腹10小時以後晨間服藥,服藥後繼續禁食2~4小時;每日2次的製劑,首劑應空腹10小時以後服藥,服藥後繼續禁食2~4小時,第二次應在餐前或餐後2小時服藥,服藥後繼續禁食2小時。每次用250ml溫開水送服,一般要求服藥1~2小時後,方可再飲水。以普通製劑爲參比製劑時,按常規用藥劑量與方法,但應與緩釋、控釋受試製劑每日總劑量相等。
3.取樣點的設計
連續服藥時間至少經過7個消除半衰期後,連續測定3天的谷濃度(cmin),以確定血藥濃度是否達穩態。取樣點最好安排在不同天的同一時間(一般清晨),以抵消時辰對藥物動力學的影響,且便於比較。達穩態後,在最後一劑量間隔內,參照單次給藥採樣時間點設計,採取足夠血樣點,測定該間隔內穩態血藥濃度一時間數據,計算有關的藥物動力學參數如峯濃度、峯時間、穩態平均血藥濃度(cav)和AUCSS等。
4.藥物動力學數據處理
(1)列出各受試者的血藥濃度一時間數據、血藥濃度平均值與標準差,列表並作圖。
(2)求出各受試者的cmax、cmin、tmax、cav、AUCSS及各參數的平均值與標準差。cmax、tmax按實測值,cmin一般按最後一劑量間隔服藥前與τ時間實測谷濃度的平均值計算,AUCSS按梯形法計算。
穩態平均血藥濃度可用下式求出:
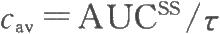
式中,AUCSS是在穩態劑量間隔期間從0~τ時間的血藥濃度一時間曲線下的面積;τ是服藥間隔時間。
(3)計算穩態時的生物利用度
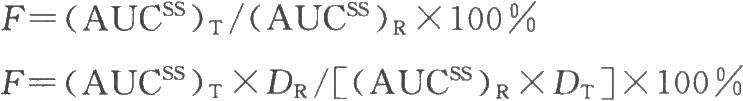
(4)血藥濃度的波動度DF(%)可用下式計算:
DF=(cmax-cmin)/cav×100%
式中,cmax爲穩態給藥期間最後一個給藥劑量的實測藥物峯濃度值;cmin爲穩態給藥期間最後一個給藥劑量實測的谷濃度。當參比製劑亦爲相同劑型的緩釋製劑時,則受試製劑的DF/τ值應不大於參比製劑DF/τ值的143%,當參比製劑爲普通製劑時,受試製劑的DF/τ值應顯著小於普通製劑。
(5)統計學分析和生物等效性評價
與普通製劑的要求相同。
4 附錄XIX C 原料藥與藥物製劑穩定性試驗指導原則
穩定性試驗的目的是考察原料藥或藥物製劑在溫度、溼度、光線的影響下隨時間變化的規律,爲藥品的生產、包裝、貯存、運輸條件提供科學依據,同時通過試驗建立藥品的有效期。
穩定性試驗的基本要求是:(1)穩定性試驗包括影響因素試驗、加速試驗與長期試驗。影響因素試驗用1批原料藥或1批製劑進行。加速試驗與長期試驗要求用3批供試品進行。(2)原料藥供試品應是一定規模生產的,供試品量相當於製劑穩定性試驗所要求的批量,原料合成工藝路線、方法、步驟應與大生產一致。藥物製劑供試品應是放大試驗的產品,其處方與工藝應與大生產一致。藥物製劑如片劑、膠囊劑,每批放大試驗的規模,片劑至少應爲10000片,膠囊劑至少應爲10000粒。大體積包裝的製劑如靜脈輸液等,每批放大規模的數量至少應爲各項試驗所需總量的10倍。特殊品種、特殊劑型所需數量,根據情況另定。(3)供試品的質量標準應與臨牀前研究及臨牀試驗和規模生產所使用的供試品質量標準一致。(4)加速試驗與長期試驗所用供試品的包裝應與上市產品一致。(5)研究藥物穩定性,要採用專屬性強、準確、精密、靈敏的藥物分析方法與有關物質(含降解產物及其他變化所生成的產物)的檢查方法,並對方法進行驗證,以保證藥物穩定性試驗結果的可靠性。在穩定性試驗中,應重視降解產物的檢查。(6)由於放大試驗比規模生產的數量要小,故申報者應承諾在獲得批准後,從放大試驗轉入規模生產時,對最初通過生產驗證的3批規模生產的產品仍需進行加速試驗與長期穩定性試驗。
本指導原則分兩部分,第一部分爲原料藥,第二部分爲藥物製劑。
4.1 一、原料藥
原料藥要進行以下試驗。
4.1.1 (一)影響因素試驗
此項試驗是在比加速試驗更激烈的條件下進行。其目的是探討藥物的固有穩定性、瞭解影響其穩定性的因素及可能的降解途徑與降解產物,爲製劑生產工藝、包裝、貯存條件和建立降解產物分析方法提供科學依據。供試品可以用1批原料藥進行,將供試品置適宜的開口容器中(如稱量瓶或培養皿),攤成≤5mm厚的薄層,疏鬆原料藥攤成≤10mm厚的薄層,進行以下試驗。當試驗結果發現降解產物有明顯的變化,應考慮其潛在的危害性,必要時應對降解產物進行定性或定量分析。
(1)高溫試驗 供試品開口置適宜的潔淨容器中,60℃溫度下放置10天,於第5天和第10天取樣,按穩定性重點考察項目進行檢測。若供試品含量低於規定限度則在40℃條件下同法進行試驗。若60℃無明顯變化,不再進行40℃試驗。
(2)高溼度試驗 供試品開口置恆溼密閉容器中,在25℃分別於相對溼度90%±5%條件下放置10天,於第5天和第10天取樣,按穩定性重點考察項目要求檢測,同時準確稱量試驗前後供試品的重量,以考察供試品的吸溼潮解性能。若吸溼增重5%以上,則在相對溼度75%±5%條件下,同法進行試驗;若吸溼增重5%以下,其他考察項目符合要求,則不再進行此項試驗。恆溼條件可在密閉容器如乾燥器下部放置飽和鹽溶液,根據不同相對溼度的要求,可以選擇NaCl飽和溶液(相對溼度75%±1%,15.5~60℃),KNO3飽和溶液(相對溼度92.5%,25℃)。
(3)強光照射試驗 供試品開口放在裝有日光燈的光照箱或其他適宜的光照裝置內,於照度爲4500lx±500lx的條件下放置10天,於第5天和第10天取樣,按穩定性重點考察項目進行檢測,特別要注意供試品的外觀變化。
關於光照裝置,建議採用定型設備“可調光照箱”,也可用光櫥,在箱中安裝日光燈數支使達到規定照度。箱中供試品臺高度可以調節,箱上方安裝抽風機以排除可能產生的熱量,箱上配有照度計,可隨時監測箱內照度,光照箱應不受自然光的干擾,並保持照度恆定,同時防止塵埃進入光照箱內。
此外,根據藥物的性質必要時可設計試驗,探討pH值與氧及其他條件對藥物穩定性的影響,並研究分解產物的分析方法。創新藥物應對分解產物的性質進行必要的分析。
4.1.2 (二)加速試驗
此項試驗是在加速條件下進行。其目的是通過加速藥物的化學或物理變化,探討藥物的穩定性,爲製劑設計、包裝、運輸、貯存提供必要的資料。供試品要求3批,按市售包裝,在溫度40℃±2℃、相對溼度75%±5%的條件下放置6個月。所用設備應能控制溫度±2℃、相對溼度±5%,並能對真實溫度與溼度進行監測。在試驗期間第1個月、2個月、3個月、6個月末分別取樣一次,按穩定性重點考察項目檢測。在上述條件下,如6個月內供試品經檢測不符合制訂的質量標準,則應在中間條件下即在溫度30℃±2℃、相對溼度65%±5%的情況下(可用Na2CrO4飽和溶液,30℃,相對溼度64.8%)進行加速試驗,時間仍爲6個月。加速試驗,建議採用隔水式電熱恆溫培養箱(20~60℃)。箱內放置具有一定相對溼度飽和鹽溶液的乾燥器,設備應能控制所需溫度,且設備內各部分溫度應該均勻,並適合長期使用。也可採用恆溼恆溫箱或其他適宜設備。
對溫度特別敏感的藥物,預計只能在冰箱中(4~8℃)保存,此種藥物的加速試驗,可在溫度25℃±2℃、相對溼度60%±10%的條件下進行,時間爲6個月。
4.1.3 (三)長期試驗
長期試驗是在接近藥物的實際貯存條件下進行,其目的是爲制定藥物的有效期提供依據。供試品3批,市售包裝,在溫度25℃±2℃,相對溼度60%±10%的條件下放置12個月,或在溫度30℃±2℃、相對溼度65%±5%的條件下放置12個月,這是從我國南方與北方氣候的差異考慮的,至於上述兩種條件選擇哪一種由研究者確定。每3個月取樣一次,分別於0個月、3個月、6個月、9個月、12個月取樣按穩定性重點考察項目進行檢測。12個月以後,仍需繼續考察,分別於18個月、24個月、36個月,取樣進行檢測。將結果與0個月比較,以確定藥物的有效期。由於實驗數據的分散性,一般應按95%可信限進行統計分析,得出合理的有效期。如3批統計分析結果差別較小,則取其平均值爲有效期,若差別較大則取其最短的爲有效期。如果數據表明,測定結果變化很小,說明藥物是很穩定的,則不作統計分析。
對溫度特別敏感的藥物,長期試驗可在溫度6℃±2℃的條件下放置12個月,按上述時間要求進行檢測,12個月以後,仍需按規定繼續考察,制訂在低溫貯存條件下的有效期。
長期試驗採用的溫度爲25℃±2℃、相對溼度爲60%±10%,或溫度30℃±2℃、相對溼度65%±5%,是根據國際氣候帶制定的。國際氣候帶見下表。
表 國際氣候帶
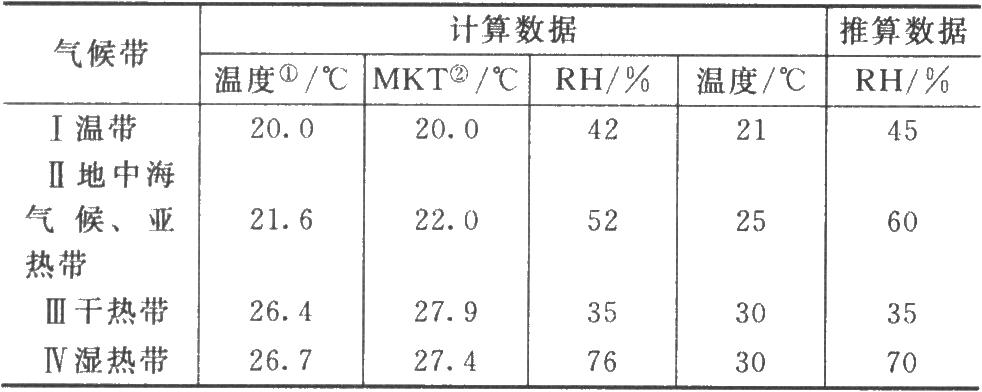
①記錄溫度;
②MKT爲平均動力學溫度。
溫帶主要有英國、北歐、加拿大、俄羅斯;亞熱帶有美國、日本、西歐(葡萄牙-希臘);乾熱帶有伊朗、伊拉克、蘇丹;溼熱帶有巴西、加納、印度尼西亞、尼加拉瓜、菲律賓。中國總體來說屬亞熱帶,部分地區屬溼熱帶,故長期試驗採用溫度爲25℃±2℃、相對溼度爲60%±10%,或溫度30℃±2℃、相對溼度65%±5%,與美、日、歐國際協調委員會(ICH)採用條件基本是一致的。
原料藥進行加速試驗與長期試驗所用包裝應採用模擬小桶,但所用材料與封裝條件應與大桶一致。
4.2 二、藥物製劑
藥物製劑穩定性研究,首先應查閱原料藥穩定性有關資料,特別瞭解溫度、溼度、光線對原料藥穩定性的影響,並在處方篩選與工藝設計過程中,根據主藥與輔料性質,參考原料藥的試驗方法,進行影響因素試驗、加速試驗與長期試驗。
4.2.1 (一)影響因素試驗
藥物製劑進行此項試驗的目的是考察製劑處方的合理性與生產工藝及包裝條件。供試品用1批進行,將供試品如片劑、膠囊劑、注射劑(注射用無菌粉末如爲西林瓶裝,不能打開瓶蓋,以保持嚴封的完整性),除去外包裝,置適宜的開口容器中,進行高溫試驗、高溼度試驗與強光照射試驗,試驗條件、方法、取樣時間與原料藥相同,重點考察項目見附表。
4.2.2 (二)加速試驗
此項試驗是在加速條件下進行,其目的是通過加速藥物製劑的化學或物理變化,探討藥物製劑的穩定性,爲處方設計、工藝改進、質量研究、包裝改進、運輸、貯存提供必要的資料。供試品要求3批,按市售包裝,在溫度40℃±2℃、相對溼度75%±5%的條件下放置6個月。所用設備應能控制溫度±2℃、相對溼度±5%,並能對真實溫度與溼度進行監測。在試驗期間第1個月、2個月、3個月、6個月末分別取樣一次,按穩定性重點考察項目檢測。在上述條件下,如6個月內供試品經檢測不符合制訂的質量標準,則應在中間條件下即在溫度30℃±2℃、相對溼度65%±5%的情況下進行加速試驗,時間仍爲6個月。溶液劑、混懸劑、乳劑、注射液等含有水性介質的製劑可不要求相對溼度。試驗所用設備與原料藥相同。
對溫度特別敏感的藥物製劑,預計只能在冰箱(4~8℃)內保存使用,此類藥物製劑的加速試驗,可在溫度25℃±2℃、相對溼度60%±10%的條件下進行,時間爲6個月。
乳劑、混懸劑、軟膏劑、乳膏劑、糊劑、凝膠劑、眼膏劑、栓劑、氣霧劑、泡騰片及泡騰顆粒宜直接採用溫度30℃±2℃、相對溼度65%±5%的條件進行試驗,其他要求與上述相同。對於包裝在半透性容器中的藥物製劑,例如低密度聚乙烯製備的輸液袋、塑料安瓿、眼用製劑容器等,則應在溫度40℃±2℃、相對溼度25%±5%的條件(可用CH3COOK·1.5H2O飽和溶液)進行試驗。
4.2.3 (三)長期試驗
長期試驗是在接近藥品的實際貯存條件下進行,其目的是爲制訂藥品的有效期提供依據。供試品3批,市售包裝,在溫度25℃±2℃、相對溼度60%±10%的條件下放置12個月,或在溫度30℃±2℃、相對溼度65%±5%的條件下放置12個月,這是從我國南方與北方氣候的差異考慮的,至於上述兩種條件選擇哪一種由研究者確定。每3個月取樣一次,分別於0個月、3個月、6個月、9個月、12個月取樣,按穩定性重點考察項目進行檢測。12個月以後,仍需繼續考察,分別於18個月、24個月、36個月取樣進行檢測。將結果與0個月比較以確定藥品的有效期。由於實測數據的分散性,一般應按95%可信限進行統計分析,得出合理的有效期。如3批統計分析結果差別較小,則取其平均值爲有效期限。若差別較大,則取其最短的爲有效期。數據表明很穩定的藥品,不作統計分析。
對溫度特別敏感的藥品,長期試驗可在溫度6℃±2℃的條件下放置12個月,按上述時間要求進行檢測,12個月以後,仍需按規定繼續考察,制訂在低溫貯存條件下的有效期。
25℃±2℃、相對溼度40%±5%,或30℃±2℃、相對溼度35%±5%的條件進行試驗,至於上述兩種條件選擇哪一種由研究者確定。
穩定性重點考察項目
原料藥及主要劑型的重點考察項目見附表,表中未列入的考察項目及劑型,可根據劑型及品種的特點制訂。
穩定性重點考察項目 | |
原料藥 | |
性狀、均勻性、含量、粒度、有關物質 | |
性狀、均勻性、含量、粒度、有關物質、分層現象 | |
性狀、均勻性、含量、粒度、有關物質 | |
如爲溶液,應考察性狀、可見異物、含量、pH值、有關物質;如爲混懸液,還應考察粒度、再分散性;洗眼劑還應考察無菌,眼丸劑應考察粒度與無菌 | |
性狀、含量、有關物質、溶散時限 | |
性狀、含量、澄清度、有關物質 |
注:有關物質(含降解產物及其他變化所生成的產物)應說明其生成產物的數目及量的變化,如有可能應說明有關物質中何者爲原料中的中間體,何者爲降解產物,穩定性試驗重點考察降解產物。
5 附錄XIX D 緩釋、控釋和遲釋製劑指導原則
緩釋、控釋製劑與普通製劑比較,藥物治療作用持久、毒副作用低、用藥次數減少。由於設計要求,藥物可緩慢地釋放進入體內,血藥濃度“峯谷”波動小,可避免超過治療血藥濃度範圍的毒副作用,又能保持在有效濃度範圍(治療窗)之內以維持療效。緩釋、控釋製劑也包括眼用、鼻腔、耳道、陰道、直腸、口腔或牙用、透皮或皮下、肌內注射及皮下植入,使藥物緩慢釋放吸收,避免門肝系統的“首過效應”的製劑。遲釋製劑係指在給藥後不立即釋放藥物的製劑,如避免藥物在胃內滅活或對胃的刺激,而延遲到腸內釋放或在結腸定位釋放的製劑,也包括在某種條件下突然釋放的脈衝製劑。
緩釋、控釋、遲釋製劑的釋藥原理主要有控制溶出、擴散、溶蝕或擴散與溶出相結合,也可利用滲透壓或離子交換機制。釋放過程可以用不同方程進行曲線擬合,如一級方程、Higuchi方程、零級方程等。緩釋與控釋的主要區別在於緩釋製劑是按時間變化先多後少地非恆速釋放,而控釋製劑是按零級速率規律釋放,即其釋藥是不受時間影響的恆速釋放,可以得到更爲平穩的血藥濃度,“峯谷”波動更小,直至基本吸收完全。通常緩釋、控釋製劑中所含的藥物量比相應一次劑量的普通製劑多,工藝也較複雜。爲了既能獲得可靠的治療效果又不致引起突然釋放(突釋)所帶來毒副作用的危險性,必須在設計、試製、生產等環節避免或減少突釋。緩釋、控釋、遲釋製劑體外、體內的釋放行爲應符合臨牀要求,且不受或少受生理與食物因素的影響。所以應有一個能反映體內基本情況的體外釋放度實驗方法,以控制製劑質量,保證製劑的安全性與有效性。
本指導原則的緩釋、控釋、遲釋製劑以口服爲重點,也可供其他給藥途徑的參考。
5.1 一、緩釋、控釋、遲釋製劑的定義
1.緩釋製劑
係指在規定釋放介質中,按要求緩慢地非恆速釋放藥物,其與相應的普通製劑比較,給藥頻率比普通製劑減少一半或給藥頻率比普通製劑有所減少,且能顯著增加患者的依從性的製劑。
2.控釋製劑
係指在規定釋放介質中,按要求緩慢地恆速釋放藥物,其與相應的普通製劑比較,給藥頻率比普通製劑減少一半或給藥頻率比普通製劑有所減少,血藥濃度比緩釋製劑更加平穩,且能顯著增加患者的依從性的製劑。
3.遲釋製劑
遲釋製劑係指在給藥後不立即釋放藥物的製劑,包括腸溶製劑、結腸定位製劑和脈衝製劑等。
腸溶製劑係指在規定的酸性介質中不釋放或幾乎不釋放藥物,而在要求的時間內,於pH6.8磷酸鹽緩衝液中大部分或全部釋放藥物的製劑。
結腸定位製劑係指在胃腸道上部基本不釋放、在結腸內大部分或全部釋放的製劑,即在規定的酸性介質與pH6.8磷酸鹽緩衝液中不釋放或幾乎不釋放,而在要求的時間內,於pH7.5~8.0磷酸鹽緩衝液中大部分或全部釋放的製劑。脈衝製劑係指不立即釋放藥物,而在某種條件下(如在體液中經過一定時間或一定pH值或某些酶作用下)一次或多次突然釋放藥物的製劑。
5.2 二、體外藥物釋放度試驗
本試驗是在模擬體內消化道條件下(如溫度、介質的pH值、攪拌速率等),對製劑進行藥物釋放速率試驗,最後制訂出合理的體外藥物釋放度,以監測產品的生產過程與對產品進行質量控制。
1.儀器裝置
除另有規定外,緩釋、控釋、遲釋製劑的體外藥物釋放度試驗可採用溶出度測定儀進行。
貼劑可採用釋放度測定法(2010年版藥典二部附錄Ⅹ D 第三法)檢查,應符合規定。
2.溫度控制
緩釋、控釋、遲釋製劑模擬體溫應控制在37℃±0.5℃,但貼劑應在32℃±0.5℃模擬表皮溫度。
3.釋放介質
以脫氣的新鮮純化水爲常用釋放介質,或根據藥物的溶解特性、處方要求、吸收部位,使用稀鹽酸(0.001~0.1mol/L)或pH3~8的磷酸鹽緩衝液,對難溶性藥物不宜採用有機溶劑,可加少量表面活性劑(如十二烷基硫酸鈉等)。
釋放介質的體積應符合漏槽條件。
除遲釋製劑外,體外釋放速率試驗應能反映出受試製劑釋藥速率的變化特徵,且能滿足統計學處理的需要,釋藥全過程的時間不應低於給藥的間隔時間,且累積釋放百分率要求達到90%以上。除另有規定外,通常將釋藥全過程的數據作累積釋放百分率一時間的釋藥曲線圖,制訂出合理的釋放度檢查方法和限度。
緩釋製劑從釋藥曲線圖中至少選出3個取樣時間點,第一點爲開始0.5~2小時的取樣時間點,用於考察藥物是否有突釋,第二點爲中間的取樣時間點,用於確定釋藥特性,最後的取樣時間點,用於考察釋藥是否基本完全。此3點可用於表徵體外緩釋製劑藥物釋放度。
控釋製劑除以上3點外,還應增加2個取樣時間點。此5點可用於表徵體外控釋製劑藥物釋放度。釋放百分率的範圍應小於緩釋製劑。如果需要,可以再增加取樣時間點。
多於一個活性成分的產品,要求對每一個活性成分均按以上要求進行釋放度測定。
5.工藝的重現性與均一性試驗
應考察3批以上、每批6片(粒)產品批與批之間體外藥物釋放度的重現性,並考察同批產品6片(粒)體外藥物釋放度的均一性。
6.釋藥模型的擬合
緩釋製劑的釋藥數據可用一級方程和Higuchi方程等擬合,即
ln(1-Mt/M∞)=-kt(一級方程)
Mt/M∞=kt1/2(Higuchi方程)控釋製劑的釋藥數據可用零級方程擬合,即Mt/M∞=kt(零級方程)
以上式中,Mt爲t時間的累積釋放量;M∞爲∞時累積釋放量;Mt/M∞爲t時累積釋放百分率。擬合時以相關係數(r)最大而均方誤差(MSE)最小的爲擬合結果最好。
5.3 三、緩釋、控釋、遲釋製劑的體內試驗
對緩釋、控釋、遲釋製劑的安全性和有效性進行評價,應通過體內的藥效學和藥動學試驗。首先對緩釋、控釋、遲釋製劑中藥物特性的物理化學性質應有充分了解,包括有關同質多晶、粒子大小及其分佈、溶解性、溶出速率、穩定性以及製劑可能遇到的其他生理環境極端條件下控制藥物釋放的變量。製劑中藥物因受處方等的影響,溶解度等物理化學特性會發生變化,應測定相關條件下的溶解特性。難溶性藥物的製劑處方中含有表面活性劑(如十二烷基硫酸鈉)時,需要瞭解其溶解特性。
關於藥物的藥動學性質,推薦採用該藥物的普通製劑(靜脈用或口服溶液,或經批准的其他普通製劑)作爲參考,對比其中藥物釋放、吸收情況,來評價緩釋、控釋、遲釋製劑的釋放、吸收情況。當設計口服緩釋、控釋、遲釋製劑時,測定藥物在胃腸道各段(尤其是當在結腸定位釋藥時的結腸段)的吸收,是很有意義的。食物的影響也應進行研究。
藥物的藥效學性質應反映出在足夠廣泛的劑量範圍內藥物濃度與臨牀響應值(治療效果或副作用)之間的關係。此外,應對血藥濃度和臨牀響應值之間的平衡時間特性進行研究。如果在藥物或藥物的代謝物與臨牀響應值之間已經有很確定的關係,緩釋、控釋、遲釋製劑的臨牀表現可以由血藥濃度一時間關係的數據表示。如果無法得到這些數據,則應進行臨牀試驗和藥動學一藥效學試驗。
緩釋、控釋、遲釋製劑進行的生物利用度與生物等效性試驗,詳見附錄XIX B。
非口服的緩釋、控釋、遲釋製劑還需對其作用部位的刺激性和(或)過敏性等進行試驗。
5.4 四、體內一體外相關性
5.4.1 (一)關於體內-體外相關性的方法
體內-體外相關性,指的是由製劑產生的生物學性質或由生物學性質衍生的參數(如tmax、cmax或AUC),與同一製劑的物理化學性質(如體外釋放行爲)之間,建立了合理的定量關係。
緩釋、控釋、遲釋製劑要求進行體內外相關性的試驗,它應反映整個體外釋放曲線與血藥濃度一時間曲線之間的關係。只有當體內外具有相關性,才能通過體外釋放曲線預測體內情況。
體內外相關性可歸納爲三種:①體外釋放曲線與體內吸收曲線(即由血藥濃度數據去卷積而得到的曲線)上對應的各個時間點應分別相關,這種相關簡稱點對點相關,表明兩條曲線可以重合。②應用統計矩分析原理建立體外釋放的平均時間與體內平均滯留時間之間的相關。由於能產生相似的平均滯留時間可有很多不同的體內曲線,因此體內平均滯留時間不能代表體內完整的血藥濃度一時間曲線。③將一個釋放時間點(t50%、t90%等)與一個藥物動力學參數(如AUC、cmax或tmax)之間單點相關,它只說明部分相關。
5.4.2 (二)本指導原則採用的方法
本指導原則緩釋、控釋、遲釋製劑體內外相關性,係指體內吸收相的吸收曲線與體外釋放曲線之間對應的各個時間點回歸,得到直線迴歸方程的相關係數符合要求,即可認爲具有相關性。
1.體內-體外相關性的建立
(1)體外累積釋放百分率-時間的體外釋放曲線
如果緩釋、控釋、遲釋製劑的釋放行爲隨外界條件變化而變化,就應該另外再製備兩種試品(一種比原製劑釋放更慢,另一種更快),研究影響其釋放快慢的外界條件,並按體外釋放度試驗的最佳條件,得到體外累積釋放百分率-時間的體外釋放曲線。
根據單劑量交叉試驗所得血藥濃度-時間曲線的數據,對在體內吸收呈現單室模型的藥物,可換算成體內吸收百分率-時間的體內吸收曲線,體內任一時間藥物的吸收百分率(Fa)可按以下Wagner-Nelson方程計算:

式中ct爲t時間的血藥濃度;
k爲由普通製劑求得的消除速率常數。
雙室模型藥物可用簡化的Loo-Riegelman方程計算各時間點的吸收百分率。
當藥物釋放爲體內藥物吸收的限速因素時,可利用線性最小二乘法迴歸原理,將同批試樣體外釋放曲線和體內吸收相吸收曲線上對應的各個時間點的釋放百分率和吸收百分率迴歸,得直線迴歸方程。
6 附錄XIX E 微囊、微球與脂質體製劑指導原則
微囊、微球、脂質體製劑係指藥物與適宜的輔料,通過微型包囊技術製得微囊、微球、脂質體,然後再按臨牀不同給藥途徑與用途製成的各種製劑。
藥物製成微囊、微球、脂質體後,可掩蓋藥物的不良氣味與口味,提高藥物的穩定性,防止藥物在胃內失活或減少對胃的刺激,可將液態藥物固態化以便運輸、應用與貯存,可減少複方藥物的配伍變化,可使製劑具有緩釋性、控釋性、遲釋性,有的還具有靶向性。
微囊、微球、脂質體可作爲藥物載體,其中具有靶向性藥物載體的製劑通常稱爲靶向製劑。靶向製劑可使藥物濃集於或接近靶細胞、靶組織、靶器官,提高療效並顯著降低對其他組織、器官及全身的毒副作用。
靶向製劑可分爲三類:①一級靶向製劑,係指進入靶部位的毛細血管牀釋藥;②二級靶向製劑,係指藥物進入靶部位的特殊細胞(如腫瘤細胞)釋藥,而不作用於正常細胞;③三級靶向製劑,係指藥物作用於細胞內的一定部位。
6.1 一、藥物載體的類型
(1)微囊係指固態或液態藥物被輔料包封成的微小膠囊。通常粒徑在1~250μm之間的稱微囊,而粒徑在0.1~1μm之間的稱亞微囊,粒徑在10~100nm之間的稱納米囊。
(2)微球係指藥物溶解或分散在輔料中形成的微小球狀實體。通常粒徑在1~250μm之間的稱微球,而粒徑在0.1~1μm之間的稱亞微球,粒徑在10~100nm之間的稱納米球。
(3)脂質體係指藥物被類脂雙分子層包封成的微小囊泡。脂質體有單室與多室之分。小單室脂質體的粒徑一般在20~80nm之間,大單室脂質體的粒徑在0.1~1μm之間,多室脂質體的粒徑在1~5μm之間。通常小單室脂質體也可稱納米脂質體。
6.2 二、常用輔料
輔料通常可分爲以下三類。
(1)在體內生物相容和可生物降解的天然材料有明膠、蛋白質、澱粉、殼聚糖、海藻酸鹽、磷脂、膽固醇等。
(2)半合成材料分爲在體內可生物降解與不可生物降解兩類。在體內可生物降解的有氫化大豆磷脂、聚乙二醇二硬脂酰磷脂酰乙醇胺等;不可生物降解的有甲基纖維素、乙基纖維素、羧甲基纖維素鹽、羥丙甲纖維素、鄰苯二甲酸乙酸纖維素等。
(3)合成材料分爲在體內可生物降解與不可生物降解兩類。可生物降解材料應用較廣的有聚乳酸、聚氨基酸、聚羥基丁酸酯、乙交酯-丙交酯共聚物等;不可生物降解的材料有聚酰胺、聚乙烯醇、丙烯酸樹脂、硅橡膠等。
此外,在製備微囊、微球、脂質體時,可加入潤溼劑、乳化劑、抗氧劑或表面活性劑等。
6.3 三、控制生產過程與貯藏期間應檢查的項目
6.3.1 (一)有害有機溶劑的限度檢查
在生產過程中引入有害有機溶劑時,應按附錄Ⅷ P殘留溶劑測定法測定,凡未規定限度者,可參考ICH,否則應制定有害有機溶劑殘留量的測定方法與限度。
6.3.2 (二)形態、粒徑及其分佈的檢查
1.形態觀察 微囊、微球、脂質體可採用光學顯微鏡觀察,粒徑小於2μm的需用掃描或透射電子顯微鏡觀察,均應提供照片。
2.粒徑及其分佈 應提供粒徑的平均值及其分佈的數據或圖形。測定粒徑有多種方法,如光學顯微鏡法、電感應法、光感應法或激光衍射法等。測定不少於500個的粒徑,由計算機軟件或下式求得算術平均徑dav。
dav=∑(nd)/∑n=(n1d1+n2d2+…+nndn)/(n1+n2+…+nn)式中,n1、n2…nn爲具有粒徑d1、d2…dn的粒子數。微囊、微球、脂質體的粒徑分佈數據,常用各粒徑範圍內的粒子數或百分率表示;有時也可用跨距表示,跨距愈小分布愈窄,即粒子大小愈均勻。
跨距=(D90-D10)/D50
式中,D10、D50、D90分別指粒徑累積分佈圖中10%、50%、90%處所對應的粒徑。
如需作圖,將所測得的粒徑分佈數據,以粒徑爲橫座標,以頻率(每一粒徑範圍的粒子個數除以粒子總數所得的百分率)爲縱座標,即得粒徑分佈直方圖;以各粒徑範圍的頻率對各粒徑範圍的平均值可作粒徑分佈曲線。
6.3.3 (三)載藥量或包封率的檢查
微囊、微球、脂質體必須提供載藥量或包封率的數據。

若得到的是分散在液體介質中的微囊、微球、脂質體,應通過適當方法(如凝膠柱色譜法、離心法或透析法)進行分離後測定,按下式計算包封率:

包封率不得低於80%。
6.3.4 (四)突釋效應或滲漏率的檢查
藥物在微囊、微球、脂質體中的情況一般有三種,即吸附、包人和嵌入。在體外釋放試驗時,表面吸附的藥物會快速釋放,稱爲突釋效應。開始0.5小時內的釋放量要求低於40%。
若微囊、微球、脂質體產品分散在液體介質中貯藏,應檢查滲漏率,可由下式計算:

6.3.5 (五)脂質體氧化程度的檢查
脂質體含有的磷脂容易被氧化,這是脂質體突出的問題。在含有不飽和脂肪酸的脂質混合物中,磷脂的氧化分三個階段:單個雙鍵的偶合、氧化產物的形成、乙醛的形成及鍵斷裂。因爲各階段產物不同,氧化程度很難用一種試驗方法評價。本指導原則採用氧化指數爲指標。
氧化指數的測定 由於氧化偶合後的磷脂在波長230nm左右具有紫外吸收峯而有別於未氧化的磷脂。測定脂質體的磷脂時,其氧化指數應控制在0.2以下。具體方法是:將磷脂溶於無水乙醇配成一定濃度的澄明溶液,分別測定在波長233nm及215nm的吸光度,由下式計算氧化指數:
氧化指數=A233nm/A215nm
6.3.6 (六)微囊、微球、脂質體製劑
應符合有關製劑通則的規定微囊、微球、脂質體製劑,除應符合本指導原則的要求外,還應分別符合有關製劑通則(如片劑、膠囊劑、注射劑、眼用製劑、鼻用製劑、貼劑、氣霧劑等)的規定。
若微囊、微球、脂質體製成緩釋、控釋、遲釋製劑,則應符合緩釋、控釋、遲釋製劑指導原則的要求。
6.3.7 (七)靶向製劑評價
7 附錄XIX F 藥品雜質分析指導原則
本附錄爲藥品質量標準中化學合成或半合成的有機原料藥及其製劑雜質分析的指導原則,供藥品研究、生產、質量標準起草和修訂參考。
任何影響藥品純度的物質均稱爲雜質。藥品質量標準中的雜質係指在按照經國家有關藥品監督管理部門依法審查批准的規定工藝和規定原輔料生產的藥品中,由其生產工藝或原輔料帶入的雜質,或在貯存過程中產生的雜質。藥品質量標準中的雜質不包括變更生產工藝或變更原輔料而產生的新的雜質,也不包括摻入或污染的外來物質。藥品生產企業變更生產工藝或原輔料,並由此帶進新的雜質對原質量標準的修訂,均應依法向有關藥品監督管理部門申報批准。藥品中不得摻入或污染藥品或其組分以外的外來物質。對於假劣藥品,必要時應根據各具體情況,可採用非法定分析方法予以檢測。
7.1 1.雜質的分類及其在藥品質量標準中的項目名稱
按化學類別和特性,雜質可分爲:有機雜質、無機雜質、有機揮發性雜質。按其來源,雜質可分爲:有關物質(包括化學反應的前體、中間體、副產物和降解產物等)、其他雜質和外來物質等。按結構關係,雜質又可分爲:其他甾體、其他生物鹼、幾何異構體、光學異構體[1]和聚合物等。按其毒性,雜質又可分爲:毒性雜質和普通雜質等。普通雜質即爲在存在量下無顯著不良生物作用的雜質,而毒性雜質爲具強烈不良生物作用的雜質。由於雜質的分類方法甚多,所以,藥品質量標準中檢查項下雜質的項目名稱,應根據國家藥典委員會編寫的《國家藥品標準工作手冊》的要求進行規範。如有機雜質的項目名稱可參考下列原則選用。
(1)檢查對象明確爲某一物質時,就以該雜質的化學名作爲項目名稱,如磷酸可待因中的“嗎啡”,氯貝丁酯中的“對氯酚”,鹽酸苯海索中的“哌啶苯丙酮”,鹽酸林可黴素中的“林可黴素B”以及胰蛋白酶中的“糜蛋白酶”等。如果該雜質的化學名太長,又無通用的簡稱,可參考螺內酯項下的“巰基化合物”、腎上腺索中的“酮體”、鹽酸地芬尼多中的“烯化合物”等,選用相宜的項目名稱。在質量標準起草說明中應寫明已明確雜質的結構式。
(2)檢查對象不能明確爲某一單一物質而又僅知爲某一類物質時,則其項目名稱可採用“其他甾體”、“其他生物鹼”、“其他氨基酸”、“還原糖”、“脂肪酸”、“芳香第一胺”、“含氯化合物”、“殘留溶劑”或“有關物質”等。
(3)未知雜質,僅根據檢測方法選用項目名稱,如“雜質吸光度”、“易氧化物”、“易炭化物”、“不揮發物”、“揮發性雜質”等。
7.2 2.質量標準中雜質檢查項目的確定
新原料藥和新制劑中的雜質,應按國家有關新藥申報要求進行研究,也可參考ICH的文本Q3A(新原料藥中的雜質)和Q3B(新制劑中的雜質)進行研究,並對雜質和降解產物進行安全性評價。新藥研製部門對在合成、純化和貯存中實際存在的雜質和潛在的雜質,應採用有效的分離分析方法進行檢測。對於表觀含量在0.1%及其以上的雜質以及表觀含量在0.1%以下的具強烈生物作用的雜質或毒性雜質,予以定性或確證其結構。對在穩定性試驗中出現的降解產物,也應按上述要求進行研究。新藥質量標準中的雜質檢查項目應包括經研究和穩定性考察檢出的,並在批量生產中出現的雜質和降解產物,幷包括相應的限度,結構已知和未知的這類雜質屬於特定雜質(specified impurities)。除降解產物和毒性雜質外,在原料中已控制的雜質,在製劑中一般不再控制。原料藥和製劑中的無機雜質,應根據其生產工藝、起始原料情況確定檢查項目,但對於毒性無機雜質,應在質量標準中規定其檢查項。
在仿製藥品的研製和生產中,如發現其雜質模式與其原始開發藥品不同或與已有法定質量標準規定不同,需增加新的雜質檢查項目的,應按上述方法進行研究,申報新的質量標準或對原質量標準進行修訂,並報有關藥品監督管理部門審批。共存的異構體和抗生素多組分一般不作爲雜質檢查項目,作爲共存物質,必要時,在質量標準中規定其比例,以保證生產用的原料藥與申報註冊時的一致性。但當共存物質爲毒性雜質時,該物質就不再認爲是共存物質。單一對映體藥物,其可能共存的其他對映體應作爲雜質檢查。消旋體藥物,當已有其單一對映體藥物的法定質量標準時,應在該消旋體藥物的質量標準中設旋光度檢查項目。單一對映體藥物,其可能共存的其他對映體應作爲雜質檢查。消旋體藥物,當已有其單一對映體藥物的法定質量標準時,應在該消旋體藥物的質量標準中設旋光度檢查項目。[1]
殘留溶劑,應根據生產工藝中所用有機溶劑及其殘留情況,確定檢查項目。可參考本藥典關於殘留溶劑的要求,或參考ICH文本Q3C(殘留溶劑指導原則)。對殘留的毒性溶劑,應規定其檢查項目。
7.3 3.雜質檢查分析方法和雜質的限度
雜質檢查分析方法應專屬、靈敏。雜質檢查應儘量採用現代分離分析手段,主成分與雜質和降解產物均能分開,其檢測限應滿足限度檢查的要求,對於需作定量檢查的雜質,方法的定量限應滿足相應的要求。
雜質檢查分析方法的建立應按本藥典的要求作方法驗證。在研究時,應採用幾種不同的分離分析方法或不同測試條件以便比對結果,選擇較佳的方法作爲質量標準的檢查方法。雜質檢查分析方法的建立,應考慮普遍適用性,所用的儀器和試材應容易獲得。對於特殊試材,應在質量標準中寫明。在雜質分析的研究階段,可用可能存在的雜質、強制降解產物,分別或加入主成分中,配製供試溶液進行色譜分析,調整色譜條件,建立適用性要求,保證方法專屬、靈敏。
新藥研究中的雜質和降解產物,或在非新藥中發現的新雜質和新降解產物,應進行分離純化製備或合成製備,以供進行安全性和質量研究。對確實無法獲得的雜質和降解產物,研製部門在申報資料和質量標準起草說明中應寫明理由。
在用現代色譜技術對雜質進行分離分析的情況下,對特定雜質中的已知雜質和毒性雜質,應使用雜質對照品進行定位;如無法獲得該對照品時,可用相對保留值進行定位;特定雜質中的未知雜質可用相對保留值進行定位。應使用多波長檢測器研究雜質在不同波長下的檢測情況,並求得在確定的一個波長下,已知雜質,特別是毒性雜質對主成分的相對響應因子。已知雜質或毒性雜質對主成分的相對響應因子在0.9~1.1範圍內時,可以用主成分的自身對照法計算含量,超出0.9~1.1範圍時,宜用對照品對照法計算含量。也可用經驗證的相對響應因子進行校正後計算。特定雜質中未知雜質的定量可用主成分自身對照品法進行計算。非特定雜質 (unspecified impurities)的限度一般爲不得超過0.10%。雜質定量計算方法應明確規定在質量標準中。一般,質量標準中還應有單個雜質限量和總雜質限量的規定。
在用薄層色譜法分析雜質時,可採用雜質對照品或主成分的梯度濃度溶液比對,對雜質斑點進行半定量評估,質量標準中應規定雜質的個數及其限度。
對於立體異構體雜質的檢測廣泛採用手性色譜法,尤其是手性高效液相色譜法,包括手性固定相法和手性流動相添加劑法(直接法)、手性試劑衍生化法(間接法),其中手性固定相法由於其一般不需衍生化、定量分析準確性高、操作簡便等特點,在手性藥物的雜質檢測中應用較多,缺點是每種固定相的適用對象有限制,需根據藥物的結構特徵選擇合適的手性柱。對於立體異構體雜質檢查方法的驗證,立體專屬性(選擇性)和手性轉化是實驗考察的重點;通常立體異構體雜質的出峯順序在前,而母體藥物在後,有利於兩者的分離和提高檢測靈敏度。另外,由於手性色譜法不能直接反映手性藥物的光學活性,需要與旋光度或比旋度測定相互補充,以有效控制手性藥物的質量。[1]
由於色譜法雜質限度檢查受色譜參數設置值的影響較大,有關操作注意事項應在起草說明中寫明,必要時,可在質量標準中予以規定。
雜質限度的制訂應考慮如下因素:雜質及含一定限量雜質的藥品的毒理學研究結果;給藥途徑;每日劑量;給藥人羣;雜質藥理學可能的研究結果;原料藥的來源;治療週期;在保證安全有效的前提下,藥品生產企業對生產高質量藥品所需成本和消費者對藥品價格的承受力。
8 附錄XIX G 正電子類放射性藥品質量控制指導原則
正電子類放射性藥品系指含有發射正電子的放射性核素的藥品。它一般由醫療機構或者正電子類放射性藥品生產企業於臨牀使用前製備。發射正電子的放射性核素主要有兩種來源:通過迴旋加速器製備和發生器製備。本指導原則僅適用於迴旋加速器製備的正電子類放射性藥品的質量控制。發生器製備的正電子類放射性藥品,參照《鍀[99mTc]放射性藥品質量控制指導原則》進行質量控制。
爲保證正電子類放射性藥品用藥安全有效,必須依據國家藥品質量標準對製備的正電子類放射性藥品進行質量控制。如果某種正電子類放射性藥品尚未有國家標準,製備單位應起草該藥品的質量標準,並經過中國藥品生物製品檢定所複覈,在確認後方可用於該藥品的質量控制。
1.發射正電子的放射性核素物理半衰期一般很短,正電子類放射性藥品的製備必須迅速。爲保證操作人員免受過量的電離輻射,一般採用自動化合成系統。
2.一般於臨用前由醫療機械自行製備和合成。鑑於氟[18F]的半衰期稍長,含氟[18F]的放射性藥品可由附近的具有正電子類放射性藥品製備資格的醫療機構或生產企業製備和供應。
3.正電子類放射性藥品批量較少,一般每批僅爲數劑。
鑑於正電子類放射性藥品製備和質量控制的特點,臨牀使用前不可能對每一批正電子類放射性藥品進行全項檢驗。爲保證正電子類放射性藥品的質量,確保用藥安全有效,規範正電子類放射性藥品的質量控制,根據《藥品管理法》和《放射性藥品管理辦法》,制訂本指導原則。
8.1 一、放射性核素的半衰期大於20分鐘的正電子類放射性藥品(如含氟[18F]的放射性藥品)
每批藥品在使用前,應對如下項目進行質量控制:
3.放射化學純度測定
4.放射性活度或濃度測定其他項目進行追溯性檢驗。
8.2 二、放射性核素的半衰期小於或等於20分鐘的正電子類放射性藥品(如含碳[11C]、氮[13N]、氧[15O]的放射性藥品)
將在同一天相同條件下製備的所有同品種製劑定義爲一批,而在一天內每次製備的製劑稱爲亞批。將在相同條件下製備的第一個亞批用於質量控制,在製備其他亞批前,至少對如下項目進行質量檢驗:
3.放射化學純度測定
4.放射性活度或濃度測定其他項目進行追溯性檢驗。
8.3 三、追溯性檢驗
正電子類放射性藥品的追溯性檢驗,應對在同一操作規範下製備的成品進行至少連續6批樣品檢驗。如結果均符合規定的則可定期進行抽驗,但至少1個月進行1次全檢。
8.4 四、檢驗結果
上述檢驗,如有一項不符合標準規定的,應立即停止製備和使用。待查明原因、合理解決,並經過3批成品驗證符合規定後,方可繼續製備。已用於臨牀的,應對患者進行跟蹤隨訪,採取必要的措施;如發生嚴重不良反應的按規定向當地藥品監督管理部門和衛生行政部門報告。
8.5 五、質量保證措施
1.製備正電子類放射性藥品的生產企業和醫療機構,應具備與製備和檢驗正電子類放射性藥品相適應的場所、儀器和設備。儀器設備應定期校驗,確保狀態正常,並有儀器設備操作和校驗規程、使用和維修記錄。
2.製備和檢驗正電子類放射性藥品的生產企業和醫療機構應具有相應專業技術人員,並經過培訓。質量控制人員應經過中國藥品生物製品檢定所或國家食品藥品監督管理局授權的機構有關放射性藥品檢驗知識的培訓,並取得培訓合格證書。
3.正電子類放射性藥品製備和檢驗應制定相應的標準操作規程,並嚴格執行。應有製備和檢驗記錄,記錄至少保存1年。
4.確保正電子類放射性藥品製備和檢驗所用原料、物料和試劑符合相關規定的品質要求;並制定原料、物料和試劑的訂購、貯存和使用管理規定。
5.爲保證自動化合成工藝的穩定,對計算機和相關自動化設備應予以控制,不得擅自改變參數。如需改變,必須經授權人員按規定進行,每次修改應予以記錄和驗證。
6.應定期對操作規程和控制工藝流程的計算機軟件進行驗證,1年至少驗證1次。如變更操作規程或計算機軟件,應進行重新驗證,並對至少連續製備的3批成品進行檢驗,結果符合質量標準規定時,方可用於正電子類放射性藥品的製備。
7.應定期對正電子類放射性藥品製備的淨化間或超淨臺的淨化性能進行驗證,確保其符合要求。
8.醫療機構首次製備的正電子類放射性藥品用於臨牀前,需連續製備3批樣品經過中國藥品生物製品檢定所或國家食品藥品監督管理局授權的藥品檢驗機構檢驗,檢驗結果符合規定後,方可進入臨牀應用。
9 附錄XIX H 鍀[99mTc]放射性藥品質量控制指導原則
鍀[ggmTc]放射性藥品系指含有放射性核素鍀[99mTc],用於臨牀診斷的藥品。它包括從鉬-鍀發生器淋洗得到的高鍀[99mTc]酸鈉注射液及利用高鍀[99mTc]酸鈉注射液和注射用配套藥盒製備得到的放射性藥品。
鍀[99mTc]放射性藥品一般由即時標記放射性藥品生產企業或具有第三類以上(包括第三類)《放射性藥品使用許可證》的醫療機構,在無菌操作條件下,以高鍀[99mTc]酸鈉注射液和相應注射用配套藥盒製備得到。鍀[99mTc]放射性藥品的製備涉及環節較多,除高鍀[99mTc]酸鈉注射液和注射用配套藥盒必須符合相應的質量標準外,對最終的成品必須進行質量檢驗。由於鍀[99mTc]的物理半衰期僅爲6.02小時,爲此,以其製備的藥品必須在製備後數十分鐘至數小時內使用,不可能在完成全部質量檢驗後才發貨或使用。根據《放射性藥品管理辦法》第十六條規定,鍀[99mTc]放射性藥品可邊檢驗邊發貨或使用。同時,一批鍀[99mTc]放射性藥品僅爲1劑或數劑藥品(一般體積僅爲數毫升),對每一批鍀[99mTc]放射性藥品進行全部質量檢驗是不現實的。
鑑於鍀[99mTc]放射性藥品的特殊性,爲了保證鍀[99mTc]放射性藥品質量及其用藥安全有效,根據《藥品管理法》和《放射性藥品管理辦法》,特制訂本指導原則。本指導原則適用於即時標記放射性藥品生產企業和自行製備鍀[99mTc]放射性藥品的醫療機構(具有第三類以上《放射性藥品使用許可證》)對鍀[99mTc]放射性藥品的質量控制。
9.1 一、發貨或使用前必須進行檢驗的質量控制項目
1.性狀
將鍀[99mTc]放射性藥品置於鉛玻璃後通過肉眼觀察,不得出現與其相應的質量標準有明顯區別的性狀(如規定爲無色澄明液體,若發現顆粒狀物質、出現渾濁或顏色變化,應停止發貨和使用)。
2.pH值
可用經過校正的精密pH試紙檢查,其pH值應在相應法定標準規定的範圍內。
3.放射化學純度
放射化學純度應按相應的質量標準規定的方法進行測定。鑑於有些檢驗方法耗時較長,爲適應快速質量控制的要求,企業或醫療機構可以採用經過驗證的快速測定方法進行測定。快速測定方法必須經過測定本單位配製的3批以上樣品,每批樣品不少於3個時間點(即製備後即刻、有效期中間點和有效期末點)的嚴格驗證,其限值不得低於標準中的限值。在日常使用過程中,應定期對該快速測定方法進行再驗證(每年至少驗證1次),確保其準確有效。
4.放射性活度
放射性活度應參照本放射性藥品檢定法(2010年版藥典二部附錄XIII)的相應規定進行測定。
5.顆粒大小
凡標準中規定有顆粒大小檢查項的鍀[99mTc]放射性藥品,在發貨或使用前應按標準或放射性藥品檢定法(2010年版藥典二部附錄XIII)項下的“顆粒細度測定法”進行檢查。顆粒大小應符合標準規定。
9.2 二、可以邊檢驗邊發貨或使用的質量控制項目
按標準方法或參照細菌內毒素檢查法(2010年版藥典二部附錄Ⅺ E)進行檢驗。含細菌內毒素量應符合規定。
2.無菌
按無菌檢查法(2010年版藥典二部附錄Ⅺ H)進行檢驗。
3.生物分佈
凡標準中規定生物分佈試驗的鍀[99mTc]放射性藥品,應按規定進行生物分佈試驗。所使用的試驗動物應符合有關規定。
4.如果上述檢驗項目有不符合標準規定的結果時,應立即停止該批鍀[99mTc]放射性藥品的製備、發貨或使用,並檢查原因。對已用於臨牀的,應對患者進行跟蹤隨訪,採取必要的預防措施,並向當地藥品監督管理部門和衛生行政主管部門報告。
5.如果有足夠的數據(連續6批以上)說明產品細菌內毒素、無菌和生物分佈試驗結果均符合規定,則細菌內毒素、無菌和生物分佈試驗可定期檢驗。間隔時間應視檢驗結果規定。
9.3 三、相應的質量保證措施
1.製備和檢驗鍀[99mTc]放射性藥品的生產企業和醫療機構,應具備相適應的環境、儀器和設備。儀器設備應定期校驗,確保狀態正常,並有儀器設備操作和校驗規程、使用記錄、維修記錄。
2.製備和檢驗含鍀[99mTc]放射性藥品的相關人員,應具備放射性藥品有關知識,並經相應的培訓。質量控制人員應經中國藥品生物製品檢定所或國家食品藥品監督管理局授權的機構有關放射性藥品檢驗知識的培訓。
3.應制定鍀[99mTc]放射性藥品製備和檢驗的標準操作規程,並嚴格按照操作規程實施各項操作。應有製備和檢驗記錄,記錄至少保存1年。
4.確保製備和檢驗含鍀[99mTc]放射性藥品所用有關原料藥和物料符合相關規定的品質要求,並制定原料藥和物料的訂購、貯存和使用管理規定。
5.定期對用於含鍀[99mTc]放射性藥品製備的淨化間或超淨臺的潔淨性能進行驗證,確保其潔淨情況符合要求。
6.對即時標記放射性藥品生產企業,在購進新的鉬-鍀發生器,用於製備含鍀[99mTc]放射性藥品之前,應對從其淋洗得到的高鍀[99mTc]酸鈉注射液按標準進行全檢(核純度項可只檢驗含鉬[99Mo]量)。如果同一廠家生產的連續多批(6批以上)鉬-鍀發生器淋洗得到的高鍀[99mTc]酸鈉注射液的細菌內毒素和無菌檢驗結果均符合規定,則從該廠家生產的鉬-鍀發生器淋洗所得高鍀[99mTc]酸鈉注射液的細菌內毒素和無菌檢查可定期進行。但每月至少對高鍀[99mTc]酸鈉注射液進行1次全檢。在注射用配套藥盒批號更換時,應對首批製備的鍀[99mTc]放射性藥品進行驗證性全檢。
10 附錄XIX J 藥物引溼性試驗指導原則
藥物的引溼性是指在一定溫度及溼度條件下該物質吸收水分能力或程度的特性。供試品爲符合藥品質量標準的固體原料藥,試驗結果可作爲選擇適宜的藥品包裝和貯存條件的參考。具體試驗方法如下:
1.取乾燥的具塞玻璃稱量瓶(外徑爲50mm,高爲15mm),於試驗前一天置於適宜的25℃±1℃恆溫乾燥器(下部放置氯化銨或硫酸銨飽和溶液)或人工氣候箱(設定溫度爲25℃±1℃,相對溼度爲80%±2%)內,精密稱定重量(m1)。
2.取供試品適量,平鋪於上述稱量瓶中,供試品厚度一般約爲1mm,精密稱定重量(m2)。
3.將稱量瓶敞口,並與瓶蓋同置於上述恆溫恆溼條件下24小時。
4.蓋好稱量瓶蓋子,精密稱定重量(m3)。

5.引溼性特徵描述與引溼性增重的界定潮解:吸收足量水分形成液體。
極具引溼性:引溼增重不小於15%。
有引溼性:引溼增重小於15%但不小於2%。
略有引溼性:引溼增重小於2%但不小於0.2%。無或幾乎無引溼性:引溼增重小於0.2%。
11 附錄XIX K 近紅外分光光度法指導原則
近紅外分光光度法系通過測定物質在近紅外光譜區(波長範圍約在780~2500nm,按波數計約爲12800~4000cm-1)的特徵光譜並利用化學計量學方法提取相關信息,對物質進行定性、定量分析的一種光譜分析技術。近紅外光譜主要由C-H、N-H、O-H和S-H等基團基頻振動的倍頻和合頻組成,由於其吸收強度遠低於物質中紅外光譜(4000~400cm-1)的基頻振動,而且吸收峯重疊嚴重,因此通常不能直接對其進行解析,而需要對測得的光譜數據進行數學處理後,才能進行定性、定量分析。
11.1 一、應用範圍
近紅外分光光度法具有快速、準確、對樣品無破壞的檢測特性,不僅能進行“離線”分析,還能直接進行“在線”過程控制;不僅可以直接測定原料和製劑中的活性成分,還能對藥品的某些理化性質如水分、脂肪類化合物的羥值、碘值和酸值等進行分析;並能對藥物輔料、中間產物以及包裝材料進行定性和分級。
11.2 二、儀器裝置
1.儀器
近紅外分光光度計由光源、單色器(或干涉儀)、採樣系統、檢測器、數據處理器和評價系統等組成。常採用高強度的石英或鎢燈光源,但鎢燈比較穩定;單色器有聲光可調型、光柵型和棱鏡型;樣品池、光纖探頭、液體透射池、積分球是常用的採樣裝置;硅、硫化鉛、砷化銦、銦鎵砷、汞鎘碲和氘代硫酸三甘肽檢測器爲常用的檢測器。檢測器和採樣系統需根據供試品的類型選擇。
2.儀器性能的校驗與自檢
爲確保儀器能達到預期的應用目的,應採用標準參比物質(SRM)對儀器的性能定期進行校驗,並在使用中通過自檢確保儀器的適用性。近紅外光譜儀的校驗參數通常包括波長的準確度、吸收/反射度的精密度、線性及最大和最小光通量處的噪聲。近紅外光譜儀的自檢通常通過比較實測光譜與校驗時儲存於儀器中的標準光譜的差異來實現。自檢時除針對上述校驗參數設計適當的指標外,還應考慮分析過程中波長的漂移和靈敏度的改變。
儀器的校驗除應定期進行外,當維修光路或更換光學部件如光源或採樣附件後也應進行。推薦用於藥物分析的近紅外光譜儀校驗參數見下表。
波長準確性 | SRM1920a②在1261、1681及1935nm處有峯 |
波長允許誤差 | 1200nm處±1nm或8300cm-1處±8cm-1 1600nm處±1nm或6250cm-1處±4cm-1 2000nm處±1.5nm或5000cm-1處±4cm-1 |
分別在1200nm、1600nm、2000nm處的 AOBS/AREF③,斜率1.0±0.05,截距0.0±0.05 | |
1200~2200nm (8300~4500cm-1)區間和100nm(300cm-1) | |
高光通量測定平均RMS | 應小於0.3×10-3,單個RMS測得值不得大於0.8×10-3 |
低光通量測定平均RMS | 應小於1×10-3,單個RMS測得值不得大於2.0×10-3 |
①通常在2500nm(4000cm-1)處儀器允許的最大漂移爲10nm(16cm-1);
②SRM1920a是美國NIST提供的用於近紅外波長校正的標準物質,通過SRM1920a對儀器1935nm處的光譜峯進行校準,來確定波長的準確性;
③AOBS指觀測的吸光度,AREF指反射標準物質在3個特定波長處的吸光度。
11.3 三、測量模式
反射模式主要用於分析固體樣品,近紅外光可穿至樣品內部1~3mm,未被吸收的近紅外光從樣品中反射出。分別測定樣品的反射光強度(I)與參比反射表面的反射光強度(Ir),其比值爲反射率R。lg(1/R)與波長或波數的函數爲近紅外光譜。
R=I/Ir
AR =lg(1/R)=lg(Ir/I)
固體樣品的顆粒大小、形狀、緊密程度及其他物理性質均會引起光譜基線的漂移,因此不是所有的固體混合物均符合比爾定律。可用數學方法減弱或消除粒度的影響。最常用的數學方法爲對光譜進行導數處理。當樣品量足夠大時,也可用多元散射校正方法處理數據。
2.透射模式
透射模式主要用於分析液體樣品,近紅外光穿過樣品,透射光強度(I)與波長或波數的函數爲近紅外光譜。測定樣品時樣品置於光源與檢測器之間的光路上,結果直接以透光率(T)或吸光度(A)表示。
T=I/I0
A=-lgT=lg(1/T) =lg(I0/I)
式中 I0爲入射光強度。
透射-反射模式爲透射與反射模式的結合,將反射鏡置樣品的後部,光源與檢測器在樣品的同側,近紅外光穿過樣品後經反射鏡返回,因此光程增加爲兩倍。
11.4 四、影響近紅外光譜的主要因素
環境溫度、樣品的光學性質、多晶型、樣品的含水量和溶劑殘留量、樣品厚度、硬度、光潔度及樣品的貯存時間等均對樣品的近紅外光譜有影響。液體樣品對環境溫度最敏感,不同晶型的樣品通常具有不同的近紅外光譜。
11.5 五、應用近紅外分光光度法進行定性、定量分析的基本要求
11.5.1 (一)定性分析
利用近紅外分光光度法進行定性分析的主要步驟包括:收集代表性樣品,測定光譜,選擇化學計量學方法對圖譜進行預處理和降維處理,建立定性分析模型,對模型進行驗證。
1.代表性樣品的選擇
選擇適宜的代表性樣品(如不同的生產工藝、物理形態、粒度發佈等)建立定性分析模型。模型中各類樣品的性質決定了模型的適用範圍。
2.圖譜預處理和降維處理
爲有效地提取有用信息,排除無效信息,在建立分類或校正模型時需要對譜圖進行數學預處理。歸一化處理常用於消除或減弱由位置或光程變化所導致的基線平移或強度變化;導數處理可以提高譜圖的分辨率,但導數處理的同時擴大了噪聲,因此常輔以平滑處理來消除噪聲;對固體樣品,採用多元散射校正(MSC)或標準正態變量變換(SNV)校正可以消除或減弱光散射引入的基線偏移。
多元近紅外光譜數據包含有大量的相關變量(共線性),建模時需要減少變量,即用一組新的不相關但包含相應信息的變量來代表所有數據的變化建立模型。常用的減少變量的方法是主成分分析(PCA)法。
3.建立定性分析模型
建立定性分析模型就是將樣品的性質與光譜的變化相關聯,用光譜的差異程度來區分樣品的性質。定性分析中常採用模式識別的方法對具有相似特徵的樣品進行分組。模式識別方法包括判別分析和聚類分析。判別分析要求對樣本的類別特徵有明確的定義,並按定義區分樣本;而聚類分析適用於僅需要對樣本進行分組而不需要預先知道這些樣品彼此間的確切關係。
4.模型的驗證
對定性分析模型,至少應進行模型的專屬性和重現性兩方面的驗證。
(1)專屬性 模型的專屬性通常用對已知樣品的鑑別正確率表示。不僅需要驗證真品的鑑別正確率,還需要用化學結構或性質上與模型中物質相近的樣品進行挑戰性驗證,證明模型能區分出這些物質。
(2)耐用性 模型的耐用性係指在不改變模型參數的情況下,考查正常操作中的微小變化對模型預測結果的影響。通常包括:
①不同操作者的影響;
②環境條件(如實驗室中的溫度、溼度變化)的影響;
③操作(如樣品在光學窗口的位置、液體探頭的測量深度、包裝狀況)的影響;
④儀器部件的更換。
11.5.2 (二)定量分析
利用近紅外分光光度法進行定量分析的主要步驟包括:收集樣品並進行檢驗,選擇代表性樣品,測定光譜,選擇化學計量學方法對圖譜進行預處理和降維處理,建立定量分析模型,對模型進行驗證。
1.代表性樣品的選擇
根據樣品的收集及檢驗情況,選擇能包括全部樣品理化性質差異的適宜數量的樣品作爲建模樣品。建模樣本的含量範圍應該寬於預測樣品的範圍,必要時可以通過加速實驗或特殊製備的方式獲得。
2.圖譜預處理和降維處理參見“定性分析”。
3.建立定量分析模型
近紅外光譜測量時一般不需要對樣品進行預處理,但測量時可受多種因素的影響,利用單波長光譜數據很難獲得準確的定量分析結果。現代近紅外光譜定量分析均利用多波長光譜數據,採用多元校正的方法,如多元線性迴歸(MLR)、主成分迴歸(PCR)、偏最小二乘迴歸(PLSR)和人工神經網絡(ANN)等建立分析模型。
近紅外分光光度法定量分析的方法學驗證與其他分析方法的要求相似。每個被驗證參數可被接受的限度範圍與該方法的應用目的有關,通常應考慮專屬性、線性、準確度、精密度和重現性。
11.6 六、近紅外模型的再驗證
當預測物質的物理性質改變,或物質的來源改變如產品的組成、生產工藝、原(輔)料的來源或級別發生改變時,需要對已建立的定量模型進行再驗證。必要時應對模型進行維護或建立新模型。
11.7 七、近紅外模型的傳遞
近紅外模型的傳遞表示模型在不同的近紅外光譜儀中的適用情況。當近紅外模型在非建模儀器中應用時,必須考慮儀器型號、數據格式、光譜範圍、數據點數量、光譜分辨率等對模型的影響。用適宜的代表性樣品(數量依據具體模型確定)分別在建模儀器(源機)和其他儀器掃描光譜,分別利用不同儀器上獲得的光譜預測結果,並進行統計學檢驗,以確證該模型在其他儀器中使用是否有效。
12 附錄XIX L 拉曼光譜法指導原則
拉曼光譜法是研究化合物分子受光照射後所產生的散射光與入射光能量差與化合物振動頻率、轉動頻率間關係的分析方法。
與紅外光譜類似,拉曼光譜是一種振動光譜技術。所不同的是,前者與分子振動時偶極矩變化相關,而拉曼效應則是分子極化率改變的結果,被測量的是非彈性的散射輻射。拉曼光譜通常採用激光作爲單色光源,將樣品分子激發到某一虛態,隨後受激分子弛豫躍遷到一個與基態不同的振動能級,此時,散射輻射的頻率將與入射頻率不同。這種頻率變化與基態和終態的振動能級差相當。這種“非彈性散射”光就稱之爲拉曼散射。頻率不變的散射稱爲彈性散射,即所謂瑞利散射。如果產生的拉曼散射頻率低於入射頻率,則稱之爲斯托克散射。反之,則稱之爲反斯托克散射。實際上,幾乎所有的拉曼分析都是測量斯托克散射。
拉曼光譜與紅外吸收光譜相似。用散射強度對拉曼位移作圖。拉曼位移(以cm-1爲單位)爲激發光的波數與散射輻射的波數之差。由於功能團或化學鍵的拉曼位移與它們在紅外光譜中的吸收波數相一致,所以譜圖的解析也與紅外吸收光譜相同。然而,通常在拉曼光譜中出現的強譜帶在紅外光譜中卻成爲弱譜帶甚至不出現,反之亦然。所以,這兩種光譜技術常互爲補充。
拉曼光譜的優點在於它的快速、準確,測量時通常不破壞樣品(固體、半固體、液體或氣體),樣品製備簡單甚至不需樣品製備。譜帶信號通常處在可見或近紅外光範圍,可以有效地和光纖聯用。這也意味着譜帶信號可以從包封在任何對激光透明的介質,如玻璃、塑料內,或將樣品溶於水中獲得。現代拉曼光譜儀使用簡單,分析速度快(幾秒到幾分鐘),性能可靠。因此,拉曼光譜與其他分析技術聯用比其他光譜聯用技術從某種意義上說更加簡便(可以使用單變量和多變量方法以及校準)。除常規的拉曼光譜外,還有一些較爲特殊的拉曼技術。它們是共振拉曼、表面增強拉曼光譜、拉曼旋光、相關-反斯托克拉曼光譜、拉曼增益或減失光譜以及超拉曼光譜等。其中,在藥物分析應用相對較多的是共振拉曼和表面增強拉曼光譜法。
共振拉曼光譜法 當激光頻率接近或等於分子的電子躍遷頻率時,可引起強烈的吸收或共振,導致分子的某些拉曼譜帶強度急劇增強數百萬倍,這就是共振拉曼效應。
許多藥物在紫外-可見光區有強的電子躍遷。某些含髮色團化合物的拉曼光譜因共振而增強,而其基體物質的光譜卻不會增強。共振拉曼技術與常規拉曼光譜技術不同之處在於要求光源可變,可調諧染料激光器是獲得共振拉曼光譜的必要條件。
有些化合物可通過化學反應改變其結構,使之最大吸收峯接近激發光頻率,如生成有色化合物,然後再進行共振拉曼光譜測定也是一個提高靈敏度的較有效的方法。
共振拉曼技術由於靈敏度高而特別適用於藥物和生物大分子的研究。缺點是由樣品本身或由雜質引起的熒光干擾,以及這一光譜技術需要特殊的激光光源和光學設計。
表面增強拉曼光譜法 吸附在極微小金屬顆粒表面或其附近的化合物(或離子)的拉曼散射要比該化合物的正常拉曼散射增加103~106倍。這種表面增強拉曼散射(SERS)在銀表面上最強,在金或銅的表面上也可觀察到。
SERS現象主要由金屬表面基質受激而使局部電磁場增強所引起。效應的強弱取決於與光波長相對應的表面粗糙度大小,以及和波長相關的複雜的金屬電介質作用的程度。許多SERS基質可以用於藥物分析,最常用的包括溶膠、電極、電介質表面金屬膜等。
帶孤對電子或π電子雲的分子呈現的SERS效應最強,其他芳氮或含氧化合物,如芳胺和酚,也具有強的SERS活性,這一效應在其他電負性功能團如羧酸中也能觀察到。從少數分子獲得大量結構信息的可能性使得SERS可用於解決高靈敏度化學分析的許多問題。在表面增強拉曼光譜中,熒光的干擾可有效地得到抑制。
12.1 1.儀器裝置
根據獲得光譜的方式,拉曼光譜儀可分爲FT拉曼光譜儀和色散型拉曼光譜儀,但所有的現代拉曼光譜儀均包括激光光源、樣品裝置、濾光系統、光波處理系統(單色器或干涉儀)和檢測器等。
(1)激光光源 下表列出幾種在藥學應用中經常使用的激光。紫外激光有時也有特殊應用,但是由於種種原因在常規分析中很少採用。
表 藥學應用中的主要激光
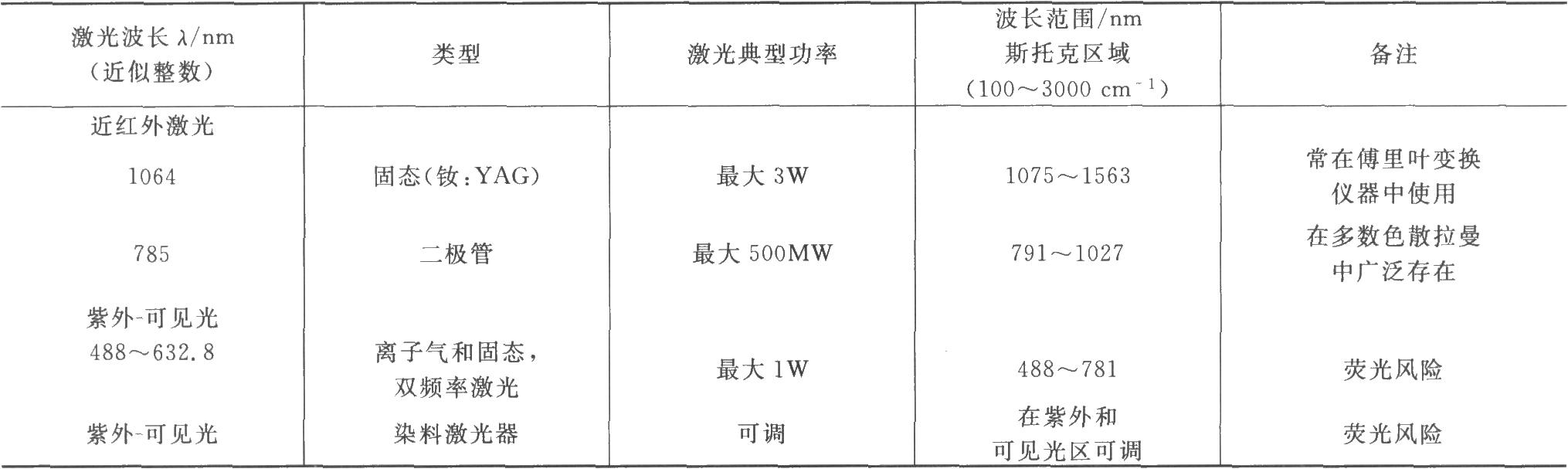
(2)樣品裝置 可有各種各樣的樣品放置方式,包括直接的光學界面、顯微鏡、光纖維探針(不接觸或光學浸入)和樣品室(包括特殊的樣品盛器和自動樣品轉換器)。樣品光路也可被設計成能獲得偏振相關拉曼光譜,這種光譜通常包含附加信息。樣品裝置的選擇應根據待測物的具體情況(如樣品的狀態、體積等)以及測量的速度、激光的安全性和樣品圖譜的質量要求等決定。
(3)濾光系統 激光波長的散射光(瑞利光)要比拉曼信號強幾個數量級,必須在進入檢測器前濾除。陷波濾波器幾乎被普遍應用於這個目的,它具有濾波效果好和體積小等優點。另外,爲防止樣品不被外輻射源(例如:房間的燈光、激光等離子體)照射,需要設置適宜的濾波器或者物理屏障。
(4)光波處理系統 光波信號可通過色散或者干涉(傅里葉變換)來處理。任何合格儀器都適用於定性鑑別。然而,選擇定量測定用儀器時,應注意色散和線性響應可能在整個波譜範圍內並不均衡(例如當使用階梯光柵分光鏡時)。
(5)檢測器 硅質CCD是色散儀器中最常用的檢測器。這種冷卻的陣列檢測器允許在低噪聲下的快速全光譜掃描。常與通常使用的785nm二極管激光器配合使用。傅里葉變換儀器通常採用單通道鍺或銦鎵砷化合物(InGaAs)檢測器以配合釹:釔-鋁-石榴紅(Nd:YAG)1064nm的激光器在近紅外區使用。
12.2 2.儀器的校正與檢定
拉曼儀器的校準包括三個要素:初始波長(X軸)、激光波長以及強度(Y軸)。
儀器供應商應該提供一種由用戶可以執行的,對儀器相關參數校正的方法,除另有規定外,使用者應根據儀器所提供的校正方法制訂具體的SOP,並嚴格按照SOP對上述參數進行檢定。
特別需要注意到,激光波長變化可影響儀器的波長精度和光度(強度)精度。即使是最穩定的激光器在使用過程中,其輸出波長也會有輕微變化。所以,激光波長必須被校正以確保拉曼位移的準確性。可以使用儀器公司提供的拉曼位移標準參考物質進行定期校正。某些儀器可以用一種拉曼內標物與初級光路分離,外在校準裝置通過散射輻射可準確地重現這一光路。
推薦使用外部參考標準對儀器進行校正。
12.3 3.樣品製備
獲得拉曼光譜可以採用下述任一物質態:結晶態、無定形、液體、氣體或等離子體。
液體能夠在玻璃管或石英管中直接測定。無定形和微晶固體也可充填入玻璃管或石英管中直接測定。爲了獲得較大的拉曼散射光強度,通常使照射在樣品上的入射光與所檢測的拉曼散射光之間的夾角爲0°、90°和180°。樣品池的放置可有多種方式。
除另有規定外,一般用作鑑別的樣品不必制樣,用作晶型、異構體限度檢查或含量測定時,供試品的製備和具體測定方法可按各品種項下的有關規定操作。
表面增強拉曼光譜和顯微拉曼光譜的測定需要某些特殊的制樣技術。爲防止樣品分解常採用的一種辦法是旋轉技術,利用特殊的裝置使激光光束的焦點和樣品的表面做相對運動,從而避免了樣品的局部過熱現象。樣品旋轉技術除能防止樣品分解外,還能提高分析的靈敏度。
12.4 4.定性鑑別
拉曼光譜可提供有關樣品分子中存在何種功能團的結構信息。所以可用於鑑別試驗和結構解析。在相同的測定條件下,繪製供試品與對照品的拉曼光譜進行比對,若兩光譜相同,即鑑別爲同一化合物。
具有多晶現象的固體藥品,由於晶型的不同,可能導致所收集供試品的光譜圖與對照品光譜圖或與標準光譜集所收載的光譜圖不一致,遇此情況,可參照紅外分光光度法鑑別的相關內容進行處理。
光譜的形狀與所用的儀器型號和性能、激發波長、樣品測定狀態及吸水程度等因素相關。因此,進行光譜比對時,應考慮各種因素可能造成的影響。
12.5 5.含量測定
拉曼譜帶的強度與待測物濃度的關係遵守比爾定律:
Iv= KlcI0
式中 Iv爲給定波長處的峯強;
K爲儀器和樣品的參數;
l爲光路長度;
I0爲激光強度。
實際工作中,光路長度被更準確地描述爲樣品體積,這是一種描述激光聚焦和採集光學的儀器變量。上述等式是拉曼定量應用的基礎。
12.6 6.影響定量測定的因素
最主要的干擾因素是熒光、樣品的熱效應和基質或樣品自身的吸收。在拉曼光譜中,熒光干擾表現爲一個典型的傾斜寬背景。因此,熒光對定量的影響主要爲基線的偏離和信噪比下降,熒光的波長和強度取決於熒光物質的種類和濃度。與拉曼散射相比,熒光通常是一種量子效率更高的過程,甚至很少量不純物質的熒光也可以導致顯著的拉曼信號降低。使用較長的波長例如785nm或1064nm的激發光可使熒光顯著減弱。然而,拉曼信號的強度與λ-4成比例,λ是激發波長。通過平衡熒光干擾、信號強度和檢測器響應可獲得最佳信噪比。測量前將樣品用激光照射一定時間,固態物質的熒光也可得以減弱。這個過程被稱爲光致漂白,它是通過降解高吸收物質實現的。光致漂白作用在液體中並不明顯,可能是由於液體樣品的流動性或熒光物質量較多所致。
樣品加熱會造成一系列的問題,例如物理狀態的改變(熔化)、晶型的轉變或樣品的燒灼。這是有色的、具強吸收或低熱傳導的小顆粒物質常出現的問題。樣品加熱的影響通常是可觀察的,表現在一定時間內拉曼光譜或樣品的表觀變化。除了減少激光通量,有許多種方法可用來降低熱效應,例如在測量過程中移動樣品或激光,或者通過熱接觸或液體浸入來改善樣品的熱傳導。
基質或樣品本身也可吸收拉曼信號。在長波傅里葉變換拉曼系統中,拉曼信號可以與近紅外的泛頻吸收重疊。這種影響與系統的光學以及樣品的形態有關。裝填和顆粒大小的差異而引起的固體散射的可變性與這種效應有關。然而,由於在拉曼光譜中樣品的有限穿透深度和相對狹窄的波長範圍,所有這些效應的大小都沒有近紅外光譜嚴重。
定量拉曼光譜與許多其他的光譜技術不同,它是單光束零背景測量。謹慎地進行樣品測定以及使用設計合理的儀器可以使這種變異減到最小,但是並不能全部消除。所以,絕對的拉曼信號強度難以準確測量。變異的潛在來源是樣品的不透明性和樣品的不均勻性、照射樣品的激光功率的變化以及光學幾何學或樣品位置的變化。這些影響可以通過能重複的或有代表性的樣品處置方式予以減小。
由於拉曼信號絕對強度的波動,使用內標是最普通和有效的減少可變性的方法。在激光照射下,加入的內標也產生拉曼光譜,選擇其一條合適的拉曼譜帶作爲參比譜帶,將樣品的分析譜帶強度與內標的參比譜帶強度進行比較(通常比較譜帶的面積或高度)。由於內標和樣品完全處於相同的實驗條件下,一些影響因素可以相互抵消。
所選擇的內標應滿足以下要求:①化學性質比較穩定,不與樣品中被測成分或其他成分發生化學反應;②內標拉曼譜帶和待測物的拉曼譜帶互不干擾;③內標應比較純,不含有被測成分或其他干擾成分。對於非水溶液,常用的內標爲四氯化碳(459cm-1);而對於水溶液,常用的內標是硝酸根離子(1050cm-1)和高氯酸根離子。對於固體樣品,有時選擇樣品中某一拉曼譜帶作爲自身對照內標譜帶。
內標方法有幾種變通選擇。可以有目的地加入一種內標,該內標應具有與待測物互不干擾的譜帶以便檢測;在溶液中,也可利用溶劑的獨特譜帶,因爲溶劑隨樣品不同將相對保持不變;另外,在製劑中,如果賦形劑量大大超過待測組分,則可以使用該賦形劑的峯;在假設激光和樣品定位的改變可同等地影響全光譜的前提下,全光譜同樣可以用作參比。
樣品測定中需考慮的重要因素還有光譜的污染。拉曼散射是一種可以被許多外源影響掩蔽的弱效應。普通的污染源包括樣品支持物(容器或基質)和周圍光線。通常,這些問題可以通過細緻的實驗方法來識別和解決。
12.7 7.測定方法驗證
對拉曼光譜方法進行驗證是必須的,至少應考察準確度、精密度等主要指標,然而,這些指標受諸多可變因素的影響,其中熒光可能是影響方法適用性的主要變量。樣品中熒光雜質的存在完全隨樣品而異。所以,方法必須能適應不同樣品體系,必須足以將雜質的影響降到最小。
檢測器的線性必須適應可能的信號水平範圍。熒光可能使信號基線比驗證時高,這時必須設法將熒光減弱或者使驗證的方法適應較高的熒光水平。這一要求對方法的精密度、檢測限(LOD)和定量限(LOQ)同樣適用,因爲基線噪聲的增加會對這些數值產生影響。
由於熒光使基線漂移可能同樣會影響定量,所以使用時,同樣需要在不同的光致漂白作用水平進行可接受的定量驗證。
必須確定激光是否對樣品造成影響。在不同激光功率和暴露時間的條件下,對樣品目視檢查和仔細審視測得的拉曼光譜測量可以確定樣品是否改變(而不是光致漂白作用)。觀察的依據是譜帶的位置、峯強和譜帶寬度是否改變或者背景強度是否有明顯的變化。
影響方法精密度的因素還包括樣品的位置。樣品的形態對於固體或液體都是非常重要的因素,在校正模型中必須嚴密控制或說明。樣品的製備方法或樣品試架的形狀可能影響測量靈敏度,而且,該靈敏度會隨着儀器的激發光和採集光學設置的不同而不同。
13 附錄XIX M 化學藥品注射劑安全性檢查法應用指導原則
本指導原則爲化學藥品注射劑臨牀使用的安全性和製劑質量可控性而定。
化學藥品注射劑安全性檢查包括異常毒性、細菌內毒素(或熱原)、降壓物質(或組胺物質)、過敏反應等項。根據處方、工藝、用法及用量等設定相應的檢查項目並進行適用性研究。
13.1 一、化學藥品(包括抗生素、生化藥品)安全性檢查項目的設定
所用原料系動物來源或微生物發酵液提取物,組分結構不清晰或有可能污染毒性雜質且又缺乏有效的理化分析方法的靜脈用注射劑,應考慮設立異常毒性檢查項。
所用原料系動物來源或微生物發酵提取物時,組分結構不清晰且有可能污染異源蛋白或未知過敏反應物質的靜脈用注射劑,如缺乏相關的理化分析方法且臨牀發現過敏反應,應考慮設立過敏反應檢查項。
所用原料系動物來源或微生物發酵提取物時,組分結構不清晰或有可能污染組胺、類組胺樣降血壓物質的靜脈用注射劑,應考慮設立降壓物質或組胺類物質檢查項。
2.肌內注射用注射劑
所用原料系動物來源或微生物發酵提取物時,組分結構不清晰或有可能污染毒性雜質且又缺乏有效的理化分析方法的肌內注射用注射劑,應考慮設立異常毒性檢查項。
所用原料系動物來源或微生物發酵提取物時,組分結構不清晰或有可能污染異源蛋白或未知過敏反應物質的肌內注射用注射劑,如缺乏相關理化分析方法且臨牀發現過敏反應,應考慮設立過敏反應檢查項。
臨牀用藥劑量較大,生產工藝易污染細菌內毒素的肌內注射用注射劑,應考慮設細菌內毒素檢查項。
3.特殊途徑的注射劑
椎管內、腹腔、眼內等特殊途徑的注射劑,其安全性檢查項目一般應符合靜脈用注射劑的要求,必要時應增加其他安全性檢查項目,如刺激性檢查、細胞毒性檢查。
注射劑輔料使用面廣,用量大,來源複雜,與藥品的安全性直接相關。在質量控制中,應根據輔料的來源、性質、用途、用法用量,配合理化分析方法,設立必要的安全性檢查項目。
5.其他
原料和生產工藝特殊的注射劑必要時應增加特殊的安全性檢查項目,如病毒檢查、細胞毒性檢查等。
所用原料系中藥提取物的注射劑,應按中藥注射劑的要求設立相關的安全性檢查項目。
13.2 二、安全性檢查方法和檢查限值確定
檢查方法和檢查限值可按以下各項目內容要求進行研究。研究確定限值後,至少應進行3批以上供試品的檢查驗證。
本法系將一定量的供試品溶液注入小鼠體內,規定時間內觀察小鼠出現的死亡情況,以判定供試品是否符合規定。供試品的不合格表明藥品中混有超過藥物本身毒性的毒性雜
檢查方法 參照異常毒性檢查法(2010年版藥典二部附錄Ⅺ C)。
設定限值前研究 參考文獻數據並經單次靜脈注射給藥確定該注射劑的急性毒性數據(LD50或LD1及其可信限)。有條件時,由多個實驗室或多種來源動物試驗求得LD50和LD1數據。注射速度0.1ml/s,觀察時間爲72小時。如其他給藥途徑或延長觀察時間,應進行相應途徑或相應觀察時間的急性毒性試驗。
設定限值 異常毒性檢查的限值應低於該注射劑本身毒性的最低致死劑量,考慮到實驗窒間差異、動物反應差異和製劑的差異,建議限值至少應小於LD1可信限下限的1/3(建議採用1/3~1/6)。如難以計算得最低致死量,可採用小於LD50可信限下限的1/4(建議採用1/4~1/8)。如半數致死量與臨牀體重劑量之比小於20可採用LD50可信限下限的1/4或LD1可信限下限的1/3。靜脈注射最大劑量0.8ml/20g仍未見毒性反應或死亡,可以此作爲檢查限值。
如對動物、給藥途徑和給藥次數、觀察指標和時間等方法和限值有特殊要求時應在品種項下另作規定。
本法系利用鱟試劑(或家兔)測定供試品所含的細菌內毒素(或熱原)的限量是否符合規定。不合格供試品在臨牀應用時可能產生熱原反應而造成嚴重的不良後果。
檢查方法 參照細菌內毒素檢查法(2010年版藥典二部附錄Ⅺ E)或熱原檢查法(2010年版藥典二部附錄Ⅺ D)。
設定限值前研究 細菌內毒素檢查應進行干擾試驗,求得最大無干擾濃度;熱原檢查應做適用性研究,求得對家兔無毒性反應、不影響正常體溫和無解熱作用劑量。
設定限值 細菌內毒素和熱原檢查的限值根據臨牀1小時內最大用藥劑量計算,細菌內毒素檢查限值按規定要求計算,由於藥物和適應症(如抗感染、抗腫瘤、心血管藥等急重病症用藥、兒童老人用藥、複合用藥、大輸液等)的不同,限值可適當嚴格,至計算值的1/3~1/2,以保證安全用藥。熱原檢查限值可參照臨牀劑量計算,一般爲人用每千克體重每小時最大供試品劑量的2~5倍,供試品注射體積每千克體重一般不少於0.5ml,不超過10ml。
細菌內毒素測定濃度應無干擾反應,熱原限值劑量應不影響正常體溫。如有干擾或影響,可在品種項下增加稀釋濃度、調節pH和滲透壓或緩慢注射等排除干擾或影響的特殊規定。
3.降壓物質檢查
本法系通過靜脈注射限值劑量供試品,觀察對麻醉貓的血壓反應,以判定供試品中所含降壓物質的限值是否符合規定。供試品的不合格表明藥品中含有限值以上的影響血壓反應的物質,臨牀用藥時可能引起急性降壓不良反應。
檢查方法 參照降壓物質檢查法(2010年版藥典二部附錄Ⅺ G)。
設定限值前研究 供試品按一定注射速度靜脈注射不同劑量後(供試品溶液與組胺對照品溶液的注射體積一般應相同,通常爲0.2~1ml/kg),觀察供試品對貓血壓反應的劑量反應關係,求得供試品降壓物質檢查符合規定的最大劑量(最大無降壓反應劑量)。
設定限值 一般以臨牀單次用藥劑量的1/5~5倍作爲降壓反應物質檢查劑量限值,急重病症用藥儘可能採用高限。特殊情況下,如供試品有一定降壓作用,則可按最大無降壓反應劑量的1/2~1/4作爲限值劑量;供試品原液靜脈注射1ml/kg劑量未見降壓反應,該劑量可作爲給藥限值。
本法系將一定濃度的供試品和組胺對照品依次注入離體豚鼠迴腸浴槽內,分別觀察出現的收縮反應幅度並加以比較,以判定供試品是否符合規定的一種方法。不合格供試品表明含有組胺和類組胺物質,在臨牀上可能引起血壓下降和類似過敏反應等嚴重的不良反應。
設定限值前研究 在確定限值前,應考察供試品對組胺對照品引起的離體豚鼠迴腸收縮反應的干擾(抑制或增強),求得最大無收縮干擾濃度。
若供試品的處方、生產工藝等任何有可能影響試驗結果的條件發生改變時,需重新進行干擾試驗。
干擾試驗 按組胺類物質檢查法,依下列順序準確注入供試品稀釋液加對照品稀釋液低劑量、對照品稀釋液低劑量、供試品稀釋液加對照品稀釋液高劑量、對照品稀釋液高劑量(dSL+T、dSL、dSH+T、dSH),重複一次,如dSL+T及dSH+T所致的反應值與dSL及dSH所致的反應值基本一致,可認爲供試品不干擾組胺物質檢查;否則該品種不適合設立組胺物質檢查項,建議設立降壓物質檢查項。同時應進行本法與降壓物質檢查法符合性的研究。
設定限值 除特殊要求外,原則上與降壓物質檢查限值一致,以臨牀單次用藥劑量的1/5~5倍量和每千克體重0.1μg組胺劑量計算注射劑含組胺類物質檢查限值,其計算公式爲:限值L=K/M,其中K值爲人每千克體重接受的組胺量(0.1μg/kg),M爲降壓物質檢查限值(mg/kg、ml/kg、IU/kg)。供試品劑量應低於最大無收縮干擾劑量。抗腫瘤藥、心血管病藥等急重病症用藥應採用高限。
本法系將一定量的供試品皮下或腹腔注射入豚鼠體內致敏,間隔一定時間後靜脈注射供試品進行激發,觀察豚鼠出現過敏反應的情況,以此判定供試品是否符合規定。供試品不合格表明注射劑含有過敏反應物質,臨牀用藥時可能使患者致敏或產生過敏反應,引起嚴重不良反應。
檢查方法 參照過敏反應檢查法(2010年版藥典二部附錄Ⅺ K)。
設定限值前研究 測定供試品對豚鼠腹腔(或皮下)和靜脈給藥的無毒性反應劑量。必要時,可採用注射劑的半成品原輔料進行致敏和激發研究,確定致敏方式和次數,在首次給藥後14、21、28天中選擇最佳激發時間。
設定限值 致敏和激發劑量應小於該途徑的急性毒性反應劑量,適當參考臨牀劑量。一般激發劑量大於致敏劑量。常用腹腔或鼠鼷部皮下注射途徑致敏,每次每隻0.5ml,1ml靜脈注射激發。如致敏劑量較小,可適當增加致敏次數,方法和限值的特殊要求應在品種項下規定。
本法系比較組胺對照品(S)與供試品(T)引起豚鼠離體迴腸收縮的程度,以判定供試品中所含組胺類物質的限度是否符合規定。
對照品溶液的製備 精密稱取磷酸組胺對照品適量,按組胺計算,加水溶解並定量稀釋製成每1ml中含1.0mg的溶液,分裝於適宜的容器內,4~8℃貯存,在確保收縮活性符合要求的前提下,可在3個月內使用。
對照品稀釋液的製備 試驗當日,精密量取組胺對照品溶液適量,用氯化鈉注射液按高、低劑量組(dSH、dSL)配成兩種濃度的稀釋液,高劑量dSH應不致使迴腸收縮達到極限,低劑量dSL所致反應值約爲高劑量的一半,調節劑量使可以重複出現。一般組胺高低劑量的終濃度爲10-6~10-8g/ml,注入體積0.2~1.0ml爲宜。
供試品溶液的製備 按品種項下規定的限量,照對照品稀釋液低劑量(dSL)製成適當的濃度。試驗時,一般供試品溶液與對照品稀釋液的注入體積應相等。
迴腸肌營養液的製備 A液:試驗當日,取氯化鈉160.0g、氯化鉀4.0g、氯化鈣(按無水物計算)2.0g、氯化鎂(按無水物計算)1.0g與磷酸氫二鈉(含12個結晶水)0.10g,加注射用水700ml使溶解,再加入注射用水適量,使成1000ml。B液:取硫酸阿托品0.5mg、碳酸氫鈉1.0g、葡萄糖(含1個結晶水)0.5g,加適量注射用水使溶解,加A液50.0ml,混合後加注射用水使成1000ml。B液應臨用前製備,並在24小時內應用。
檢查法 取健康合格的成年豚鼠,雌雄均可,雌者無孕,體重250~350g,禁食24小時,迅速處死,將血排淨。立即剖腹取出迴腸一段(選用遠端腸段,該段最敏感),注意避免因牽拉使迴腸受損,必要時仔細分離腸繫膜。剪取2~3cm長,用注射器抽取上述溶液B,小心沖洗出腸段的內容物。將腸段下端固定於離體器官恆溫水浴裝置的浴槽底部,上端用線與肌張力換能器相連;浴槽中事先放入一定量的B液(約10~30ml),通入95% 02和5% CO2的混合氣體,維持恆溫(34~360℃),用適當方法記錄迴腸收縮曲線。如果使用槓桿,其長度應能使腸段的收縮放大約20倍。選擇lg的預負荷,可根據其靈敏度加以調節。迴腸放入浴槽後,靜置15~30分鐘,方可開始注入藥液。每次注入藥液前,要用B液沖洗浴槽3次(最好是溢出,而不排空浴槽)。相鄰兩次給藥的間隔時間應一致(約2分鐘),每次給藥前應在前一次反應恢復穩定後進行。
取對照品稀釋液和供試品溶液,照下列次序準確注入浴槽:dSL、dT、dT、dSL、dSH,如dSH所致的反應值大於dSL所致反應值時判定試驗有效。如供試品溶液引起迴腸收縮,則分別計算dSL、dT所致反應的平均值。若dT所致反應的均值不大於dSL所致反應的均值,即判定供試品組胺類物質檢查符合規定;若dT所致反應的均值大於dSL所致反應的均值,即判定供試品組胺類物質檢查不符合規定。如供試品不引起迴腸收縮,則在供試品溶液中加入組胺對照品高、低劑量,並按下列次序準確注入dSH、dSL+T、dSH+T、dSL,重複一次,若供試品組胺溶液產生的收縮與對應組胺對照液高、低劑量的收縮基本一致,可判定供試品組胺類物質檢查符合規定;若供試品組胺溶液產生的收縮與對應組胺對照液高、低劑量產生的收縮不相符,即減少或無收縮,或不能重複出現,則此檢查結果無效,應進行供試品的降壓物質檢查。
14 附錄XIX N 抑菌劑效力檢查法指導原則
抑菌劑效力檢查法系用於測定滅菌、非滅菌製劑中抑菌劑的活性,以評價最終產品的抑菌效力,同時也可用於指導生產企業在研發階段製劑中抑菌劑濃度的確定。
如果藥物本身不具有充分的抗菌活性,那麼應根據製劑特性(如水溶液製劑)添加適宜的抑菌劑,以防止製劑在正常貯藏和使用過程中可能發生的微生物污染和繁殖使藥物發生變質而對使用者造成危害,尤其是多劑量包裝的製劑。
在藥品生產過程中,抑菌劑不能用於替代藥品生產的GMP管理,不能作爲非滅菌製劑降低微生物污染的唯一途徑,也不能作爲控制多劑量包裝製劑滅菌前的生物負載的手段。所有抑菌劑都具有一定的毒性,製劑中抑菌劑的量應爲最低有效量。同時,爲保證用藥安全,最終包裝容器中的抑菌劑有效濃度應低於對人體有害的濃度。
在製劑通則中要求具有抗菌活性的製劑,不管是添加的抑菌劑,還是藥物本身具有抗菌活性,在藥物研發階段,均應確認其抗菌效力。抑菌劑的抗菌效力在貯存過程中有可能因藥物的成分或包裝容器等因素影響而提高或降低,因此,應驗證最終容器中的抑菌劑效力在效期內不因貯藏條件而降低。本試驗方法和抑菌劑抑菌效力判斷標準用於包裝未啓開的成品製劑。
14.1 產品分類
進行本試驗的藥品分爲四類(表1),以便標準的制定和執行。
14.2 培養基
培養基的製備
酪蛋白腖17.0g、磷酸二氫鉀2.5g、大豆木瓜蛋白酶消化物3.0g、氯化鈉5.0g、葡萄糖2.5g、水1000ml除葡萄糖外,取上述成分混合,微溫溶解,調pH值約7.0,煮沸,加入葡萄糖溶解後,搖勻,濾清,調節pH值使滅菌後爲7.3±0.2,分裝,滅菌。
胰酪腖15.0g、氯化鈉5.0g、大豆木瓜蛋白酶水解物5.0g、瓊脂15.0g、純化水1000ml
除瓊脂外,取上述成分,混合,微溫溶解,調節pH值使滅菌後在25℃的pH值爲7.3±0.2,加入瓊脂,加熱溶化後,搖勻,分裝,滅菌。[2]
3.沙氏葡萄糖液體培養基、沙氏葡萄糖瓊脂培養基照微生物限度檢查法(2010年版藥典二部附錄Ⅺ J)製備。
抑菌劑效力測定用培養基應進行培養基的適用性檢查,包括成品培養基、由脫水培養基或按處方配製的培養基均應檢查。
菌種 試驗所用的菌株傳代次數不得超過5代(從菌種保藏中心獲得的冷凍乾燥菌種爲第0代),並採用適宜的菌種保藏技術進行保存,以保證試驗菌株的生物學特性。
銅綠假單胞菌(Pseudmnonas aeruginosa)[CMCC(B)10 104]
大腸埃希菌(Escherichia coli)[CMCC(B) 44 102]
金黃色葡萄球菌(Staphylococcus aureus)[CMCC(B) 26 003]
白色念珠菌(Candida albicans)[CMCC(F) 98 001]
黑麴黴(Aspergillus niger)[CMCC(F) 98 003]
菌液製備 接種大腸埃希菌、金黃色葡萄球菌、銅綠假單胞菌的新鮮培養物至胰酪腖大豆肉湯培養基中,30~35℃培養18~24小時;接種白色念珠菌的新鮮培養物至沙氏葡萄糖液體培養基中,20~25℃培養24~48小時。上述培養物用0.9%無菌氯化鈉溶液製成每1ml含菌數爲50~100cfu的菌懸液。接種黑麴黴的新鮮培養物至沙氏葡萄糖瓊脂斜面培養基中,20~25℃培養5~7天,加入3~5ml含0.05% (ml/ml)聚山梨酯80的0.9%無菌氯化鈉溶液,將孢子洗脫。然後,採用適宜方法吸出孢子懸液至無菌試管內,用含0.05%(ml/ml)聚山梨酯80的0.9%無菌氯化鈉溶液製成每1ml含孢子數50~100cfu的孢子懸液。
菌液製備後若在室溫下放置,應在2小時內使用;若保存在2~8℃,可在24小時內使用。黑麴黴孢子懸液可保存在2~8℃,在驗證過的貯存期內使用。
適用性檢查 取大腸埃希菌、金黃色葡萄球菌、銅綠假單胞菌各50~100cfu,分別注入無菌平皿中,立即傾注胰酪腖大豆瓊脂培養基,每株試驗菌平行製備2個平皿,混勻,凝固,置30~35℃培養48小時,計數;取白色念珠菌、黑麴黴各50~100cfu,分別注入無菌平皿中,立即傾注沙氏葡萄糖瓊脂培養基,每株試驗菌平行製備2個平皿,混勻,凝固,置20~25℃培養72小時,計數;同時,用對應的對照培養基替代被檢培養基進行上述試驗。
結果判定 若被檢培養基上的菌落平均數不小於對照培養基上菌落平均數的70%,且菌落形態大小與對照培養基上的菌落一致,判該培養基的適用性檢查符合規定。
14.3 抑菌劑效力測定
菌種 同培養基的適用性檢查,若需要,製劑中常見的污染微生物也可作爲試驗菌株。
菌液製備 接種銅綠假單胞菌、金黃色葡萄球菌、大腸埃希菌的新鮮培養物至胰酪腖大豆肉湯培養基或胰酪腖大豆瓊脂培養基中,30~35℃培養18~24小時;接種白色念珠菌於沙氏葡萄糖液體培養基或沙氏葡萄糖瓊脂培養基中,20~25℃培養24~48小時。若爲瓊脂培養物,加入適量的0.9%無菌氯化鈉溶液將瓊脂表面的培養物洗脫,然後,用適宜方法吸出菌懸液至無菌試管內,加入適量的0.9%無菌氯化鈉溶液並採用比濁法製成每1ml含茵數約爲108cfu的菌懸液。若爲液體培養物,用離心法收集菌體,並用0.9%無菌氯化鈉溶液沖洗,採用比濁法製成每1ml含菌數約爲108cfu的菌懸液。接種黑麴黴的新鮮培養物至沙氏葡萄糖瓊脂培養基中,23~28℃培養5~7天,加入3~5ml含0.05%(ml/ml)聚山梨酯80的0.9%無菌氯化鈉溶液,將孢子洗脫,然後,用適宜方法吸出孢子懸液至無菌試管內,加入適量的含0.05%(ml/ml)聚山梨酯80的0.9%無菌氯化鈉溶液並採用比濁法製成每1ml含孢子數108cfu的孢子懸液。同時採用平皿法測定1ml菌懸液的菌數。
菌液製備後若在室溫下放置,應在2小時內使用;若保存在2~8℃,可在24小時內使用。黑麴黴的孢子懸液可保存在2~8℃,在1周內使用。
供試品接種 抑菌劑效力可能受試驗用容器特徵的影響,如容器的材質、形狀、體積及封口的方式等。因此,只要供試品每個包裝容器的裝量足夠試驗用,同時容器便於按無菌操作技術接入試驗菌液、混合及取樣等,一般應將試驗菌直接接種於供試品原包裝容器中進行貯存。若因供試品的性狀或每個容器裝量等因素需將供試品轉移至無菌容器時,該容器的材質不得影響供試品的特性(如吸附作用),特別應注意不得影響供試品的pH,pH對抑菌劑的活性影響很大,同時容器的口徑大小應便於供試品的進出及混勻。
取包裝完整的供試品至少5份,直接接種試驗菌,或取適量供試品分別轉移至5個適宜的無菌容器中(若試驗菌株數超過5株,應增加相應的供試品份數),每一容器接種一種試驗菌,1、2、3類供試品中1g或1ml接種菌量爲105~106 cfu,4類供試品中1g或1ml接種菌量爲103~104 cfu,接種菌液的體積不得超過供試品體積的0.5%~1%,充分混合,使供試品中的試驗菌均勻分佈。然後將接種的供試品在試驗期間置20~25℃,避光貯存,貯存溫度的變化應儘可能控制在最小範圍,並防止被污染。
存活菌數測定 根據產品類型,在供試品剛接種(0時)及表2規定的間隔時間,分別從上述每個容器中取供試品1ml(g),用pH7.0無菌氯化鈉-蛋白腖緩衝液稀釋成1: 10、1:102、1: 103等稀釋級。採用平皿法或薄膜過濾法(照附錄Ⅺ J微生物限度檢查法,其中測定細菌用胰酪腖大豆瓊脂培養基,測定真菌用沙氏葡萄糖瓊脂培養基)測定每份供試品中所含的菌數。菌數測定方法應進行驗證,驗證方法按微生物限度檢查法(2010年版藥典二部附錄Ⅺ J)中的“計數方法的驗證”進行,其中測定細菌用胰酪腖大豆瓊脂培養基,測定真菌用沙氏葡萄糖瓊脂培養基。
根據菌數測定結果,計算1ml(g)供試品各試驗菌所加的菌數及各間隔時間的菌數,並換算成lg值。
結果判斷 抑菌劑效力根據各間隔時間的菌數lg值相對於初始值(0時菌數lg值)減少程度進行評價(表2),試驗結果按有效數字的修約規則進舍,保留小數點後1位有效數字。結果符合表2要求可判定該產品抑菌效力符合規定。
1類供試品 | |
7天菌數下降不少於1.0 lg,14天菌數下降不少於3.0 lg,14天到28天菌數不增加 | |
與初始值比,7、14、28天菌數均不增加 | |
2類供試品 | |
14天菌數下降不少於2.0 lg.14天到28天菌數不增加 | |
與初始值比,14、28天菌數均不增加 | |
3類供試品 | |
14天菌數下降不少於1.0 lg,14天到28天菌數不增加 | |
與初始值比,14、28天菌數均不增加 | |
4類供試品 | |
與初始值比,14、28天菌數均不增加 | |
注:表中“不增加”是指對前一個測定時間,試驗菌增加的數量不超過0.5 lg。
15 附錄XIX O 藥品微生物檢驗替代方法驗證指導原則
本指導原則是爲所採用的試驗方法能否替代藥典規定的方法用於藥品微生物的檢驗提供指導。
隨着微生物學的迅速發展,製藥領域不斷引入了一些新的微生物檢驗技術,大體可分爲三類:(1)基於微生物生長信息的檢驗技術,如生物發光技術、電化學技術、比濁法等;(2)直接測定被測介質中活微生物的檢驗技術,如固相細胞技術法、流式細胞計數法等;(3)基於微生物細胞所含有特定組成成分的分析技術,如脂肪酸測定技術、核酸擴增技術、基因指紋分析技術等。這些方法與傳統檢查方法比較,或簡便快速,或具有實時或近實時監控的潛力,使生產早期採取糾正措施及監控和指導優良生產成爲可能,同時新技術的使用也促進了生產成本降低及檢驗水平的提高。
在控制藥品微生物質量中,微生物實驗室出於各種原因如成本、生產量、快速簡便及提高藥品質量等需要而採用非藥典規定的檢驗方法(即替代方法)時,應進行替代方法的驗證,確認其應用效果優於或等同於藥典的方法。
15.1 微生物檢驗的類型及驗證參數
藥品微生物檢驗方法主要分兩種類型:定性試驗和定量試驗。定性試驗就是測定樣品中是否存在活的微生物,如無菌檢查及控制菌檢查。定量試驗就是測定樣品中存在的微生物數量,如菌落計數試驗。
由於生物試驗的特殊性,如微生物檢驗方法中的抽樣誤差、稀釋誤差、操作誤差、培養誤差和計數誤差都會對檢驗結果造成影響,因此,藥品質量標準分析方法驗證指導原則(2010年版藥典二部附錄XIX A)不完全適宜於微生物替代方法的驗證。藥品微生物檢驗替代方法的驗證參數見表1。
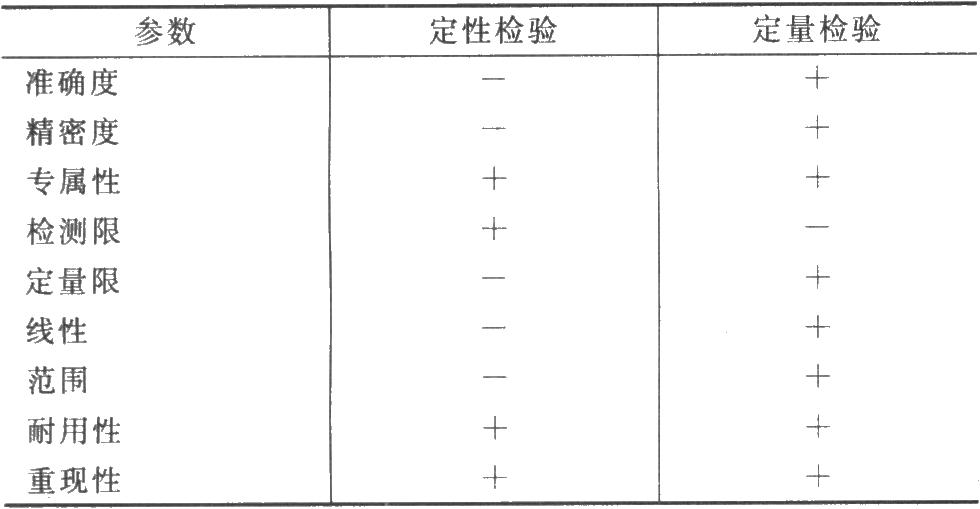
儘管替代方法的驗證參數與藥品質量標準分析方法驗證參數有相似之處,但是其具體的內容是依據微生物檢驗特點而設立的。替代方法驗證的實驗結果需進行統計分析,當替代方法屬於定性檢驗時,一般採用非參數的統計技術;當替代方法屬於定量檢驗時,需要採用參數統計技術。
進行微生物替代方法的驗證時,若替代方法只是針對藥典方法中的某一環節進行技術修改,此時,需要驗證的對象僅是該項替代技術而不是整個檢驗方法。如無菌試驗若改爲使用含培養基的過濾器,然後通過適宜的技術確認活的微生物存在,那麼,驗證時僅需驗證所用的微生物回收系統而不是整個無菌試驗方法。
15.2 替代方法驗證的一般要求
在開展替代方法對樣品檢驗的適用性驗證前,有必要對替代方法有一個全面的瞭解。首先,所選用的替代方法應具備必要的方法適用性證據,表明在不含樣品的情況下,替代方法在不同類型的微生物檢驗中所具有的專屬性、精密度和檢測限等參數。這些證據或由替代方法的研發者提供,或由方法使用者完成。
使用者在基本確認替代方法的適用性後,應採用樣品按表1規定的參數逐一進行驗證,以確認替代方法可否用於該樣品的檢驗。驗證至少使用2個批號的樣品,每批樣品應平行進行至少3次獨立實驗。
在開展各參數驗證時,涉及的菌種除應包括微生物限度檢查法(2010年版藥典二部附錄Ⅺ J)和無菌檢查法(2010年版藥典二部附錄Ⅺ H)中培養基適用性檢查規定的菌株外,還應根據替代方法及樣品的特點增加相應的菌株。各菌種應分別進行驗證。
15.3 樣品中微生物定性檢驗方法的驗證
1.專屬性
微生物定性檢驗的專屬性是指檢測樣品中可能存在的特定微生物種類的能力。當替代方法以微生物生長作爲判斷微生物是否存在時,其專屬性驗證時應確認所用培養基的促生長試驗,還應考慮樣品的存在對檢驗結果的影響。當替代方法不是以微生物生長作爲判斷指標時,其專屬性驗證應確認檢測系統中的外來成分不得干擾試驗而影響結果,如確認樣品的存在不會對檢驗結果造成影響。採用替代方法進行控制菌的檢驗,還應選擇與控制菌具有類似特性的菌株作爲驗證對象。
2.檢測限
微生物定性檢驗的檢測限是指在替代方法設定的檢驗條件下,樣品中能被檢出的微生物的最低數量。由於微生物所具有的特殊性質,檢測限是指在稀釋或培養之前初始樣品所含有的微生物數量,而不是指檢驗過程中某一環節的供試液中所含有的微生物數量。例如,微生物限度檢查中規定不得檢出沙門菌,對檢測限而言,是指每10g樣品中能被檢出的沙門菌的最低數量。
檢測限確定的方法是在樣品中接種較低濃度的試驗菌(每單位不超過5cfu),然後分別採用藥典方法和替代方法對該試驗菌進行檢驗,以檢出與否來比較兩種方法的差異。試驗菌的接種量鬚根據試驗而定,以接種後採用藥典方法50%的樣品可檢出該試驗菌爲宜。檢測限驗證至少應重複進行5次。對於同一種試驗菌可採用卡方檢驗(X2)來評價兩種方法的檢測限是否存在差異。
3.重現性
微生物定性檢驗的重現性是指相同的樣品在正常的實驗條件(如實驗地點、實驗人員、儀器、試劑的批次等)發生變化時,所得檢驗結果的精密度。重現性可視爲微生物檢驗方法在檢驗結果上抵抗操作和環境變化的能力。方法使用者應優先測定該驗證參數。在樣品中接種一定數量的試驗菌(接種量應在檢測限以上),採用藥典方法和替代方法,分別由不同人員,在不同時間,使用不同的試劑(或儀器)進行檢驗,採用卡方檢驗(X2)來評價兩種方法的重現性是否存在差異。驗證過程中,應關注樣品的一致性。
4.耐用性
微生物定性檢驗的耐用性是指當方法參數有小的刻意變化時,檢驗結果不受影響的能力,爲方法正常使用時的可靠性提供依據。方法使用者應優先測定該驗證參數。與藥典方法比較,若替代方法檢驗條件較爲苛刻,則應在方法中加以說明。替代方法與藥典方法的耐用性比較不是必須的,但應單獨對替代方法的耐用性進行評價,以便使用者瞭解方法的關鍵操作點。
15.4 樣品中微生物定量檢驗方法的驗證
微生物定量檢驗一般都涉及菌落計數。對計數結果進行數據處理時通常需要使用統計的方法。由於菌落計數服從泊松分佈,因此採用泊松分佈的統計方法對計數結果進行數據處理優於採用正態分佈的統計方法。檢驗者往往習慣採用正態分佈的統計方法,因此也可以通過對數轉換或平方根加1的方法將原始數據轉換爲正態分佈數據後再進行統計分析。兩種統計方法都適用於微生物數據的統計分析。
1.準確度
微生物定量檢驗的準確度是指替代方法的檢驗結果與藥典方法檢驗結果一致的程度。準確度的確認應在檢測的範圍內,通常用微生物的回收率(%)來表示。
檢測範圍內的準確度都應符合要求,準確度驗證的方法是:製備試驗菌的菌懸液,菌懸液的濃度應選擇爲能夠準確計數的最高濃度,然後系列稀釋至較低濃度(如小於10 cfu/ml)。例如,菌落計數平皿法的替代方法,在製備高濃度菌懸液時,其濃度可以是103cfu/ml,並系列稀釋至100 cfu/ml。每個試驗菌應至少選擇5個菌濃度進行準確度確認,替代方法的檢驗結果不得少於藥典方法檢驗結果的70%,也可以採用合適的統計學方法表明替代方法的回收率至少與藥典方法一致。當替代方法的回收率高於藥典方法時,有必要結合專屬性項下的有關內容對準確度進行評價。
2.精密度
微生物定量檢驗的精密度是指在檢驗範圍內,對同一個均勻的樣品多次重複取樣測定,其檢驗結果的一致程度,通常採用標準偏差或相對標準偏差來表示,也可以採用其他適宜的方式。
精密度驗證的方法是:製備試驗菌的菌懸液,菌懸液的濃度應選擇爲能夠準確讀數的最高濃度,然後系列稀釋至較低濃度(如小於10 cfu/ml)。每個試驗菌選擇其中至少5個濃度的菌懸液進行檢驗。每一個濃度至少應進行10次重複檢驗,以便能夠採用統計分析方法得到標準偏差或相對標準偏差。一般情況下,可以接受的相對標準偏差(RSD)應不大於35%。不考慮特殊的檢驗結果,替代方法的相對標準偏差(RSD)應不大於藥典方法。例如,藥典菌落計數平皿法其可接受的相對標準偏差(RSD)與含菌濃度的關係見表2。
表2 不同含菌濃度下預期的相對標準偏差
cfu/皿 | 預期RSD |
<10 | <35% |
10~30 | <25% |
30~300 | <15% |
3.專屬性
微生物定量檢驗的專屬性是指通過檢測適宜的試驗菌,以證明檢驗方法與其設定目的相適應的能力。例如,菌落計數平皿法其設定目的在於檢出一定數量的微生物,則其專屬性驗證應證明當樣品中存在一定數量的試驗菌時,通過平皿法檢驗,能夠檢出試驗菌,而樣品的存在不會對結果造成影響。專屬性驗證時,應能夠設計出可能使替代方法出現假陽性的實驗模型來挑戰替代方法,從而確認替代方法的適用性。當替代方法不依賴微生物生長出菌落或出現混濁就可以定量時(如不需要增菌或在1~50cfu範圍內就可直接測定菌數的定量方法),以上驗證方式就顯得更爲重要。
4.定量限
微生物定量檢驗的定量限是指樣品中能被準確定量測定的微生物最低數量。由於無法得到含有已知微生物數量的實驗樣品,因此,在定量限驗證時,應選擇在檢驗範圍內至少5個菌濃度,每個濃度重複取樣測定不少於5次,替代方法的定量限不得大於藥典方法。需要注意的是,由於細菌計數和菌落數服從泊松分佈,可能存在計數結果的誤差,因此替代方法的定量限僅需證實在相近的低限度下其靈敏度至少相當於藥典方法。
定量限驗證的方法是:在檢驗範圍的低限制備5份不同含菌濃度的菌懸液,每份菌懸液分別用藥典方法和替代方法進行不少於5次檢驗,採用統計方法比較替代方法的檢驗結果與藥典方法結果的差異,從而評價替代方法的定量限。
5.線性
微生物定量檢驗的線性是指在一定範圍內,檢驗結果與樣品中微生物數量成比例關係的程度。線性驗證時必須覆蓋能夠準確測定的所有濃度範圍。每株試驗菌應選擇至少5個濃度,每個濃度至少測定5次。根據以上實驗數據,以檢驗結果爲因變量,以樣品中微生物的預期數量爲自變量進行線性迴歸分析,計算相關係數r。當相關係數不能準確評估線性時,只能確定簡單大約的關係值。替代方法的相關係數不得低於0.95。
6.範圍
微生物定量檢驗的範圍是指能夠達到一定的準確度、精密度和線性,檢驗方法適用的高低限濃度或數量的區間。
7.重現性
微生物定量檢驗的重現性是指相同的樣品在正常的實驗條件(如實驗地點、實驗人員、儀器、試劑的批次等)發生變化時,所得檢驗結果的精密度。重現性可視爲微生物檢驗方法在檢驗結果上抵抗操作和環境變化的能力。方法使用者應優先測定該驗證參數。在樣品中接種一定數量的試驗菌(接種量應在定量限以上),採用藥典方法和替代方法,分別由不同人員,在不同時間,使用不同的試劑(或儀器)進行檢驗,對榆驗結果進行統計分析,以相對標準偏差(RSD)來評價兩種方法的重現性差異。驗證過程中,應關注樣品的一致性。
8.耐用性
微生物定量檢驗的耐用性是指當方法參數有小的刻意變化時,檢驗結果不受影響的能力,爲方法正常使用時的可靠性提供依據。方法使用者應優先測定該驗證參數。與藥典方法比較,若替代方法檢驗條件較爲苛刻,則應在方法中加以說明。替代方法與藥典方法的耐用性比較不是必須的,但應單獨對替代方法的耐用性進行評價,以便使用者瞭解方法的關鍵操作點。
16 附錄XIX P 微生物限度檢查法應用指導原則
爲更好應用微生物限度檢查法(2010年版藥典二部附錄Ⅺ J),特制定本指導原則。
微生物限度檢查法可用於判斷非規定滅菌製劑及原料、輔料是否符合藥典的規定,也可用於指導製劑、原料、輔料的微生物質量標準的制定,及指導生產過程中間產品微生物質量的監控。本指導原則將對標準和方法中的特定內容及標準的應用做進一步的說明。
1.微生物限度檢查過程中,如需要使用表面活性劑、滅活劑及中和劑,在確定其能否適用於所檢樣品及其用量時,除應證明該試劑對所檢樣品的處理有效外,還須確認該試劑不影響樣品中可能污染的微生物的檢出(即無毒性),因此無毒性確認試驗的菌株不能僅侷限於驗證試驗菌株,而應當包括產品中可能污染的微生物。
2.供試液製備方法、抑菌成分的消除方法及細菌、黴菌及酵母菌計數方法應儘量選擇微生物限度檢查法中操作簡便、快速的方法,同時,所選用的方法應避免損傷供試品中污染的微生物。對於抑菌作用較強的供試品,在供試品溶液性狀允許的情況下,應儘量選用薄膜過濾法進行試驗。
3.微生物限度檢查法(2010年版藥典二部附錄Ⅺ J)收載的離心沉澱法僅適用於製備細菌計數或控制菌(細菌)檢查用的供試液,規定的500轉/分鐘、不超過3分鐘只用於去除供試液中的沉澱物。採用該方法時,供試液中的樣品顆粒大小、黏稠度及污染的微生物大小、轉速等直接影響着樣品中微生物的回收,易造成檢驗結果不能真實反映供試品的污染情況。因此,供試液製備時儘量避免使用該方法,更不宜採用高速離心沉降集菌。
4.對照培養基係指按培養基處方特別製備、質量優良的培養基,用於培養基適用性檢查。由中國藥品生物製品檢定所研製及分發。
5.進行驗證試驗時,若因沒有適宜的方法消除供試品中的抑菌作用而導致微生物回收的失敗,應採用能使微生物生長的更高稀釋級供試液進行方法驗證試驗。此時更高稀釋級供試液的確認要從低往高的稀釋級進行,但最高稀釋級供試液選擇應根據供試品應符合的微生物限度標準和菌數報告規則,如供試品應符合的微生物限度標準是每1g供試品細菌數不得過1000cfu,那麼最高稀釋級是1:10-3。
若採用允許的最高稀釋級供試液進行驗證試驗還存在1株或多株試驗菌的回收率達不到要求,那麼應選擇回收情況最接近要求的方法進行供試品的檢測。如某種產品對某試驗
菌有較強的抑菌性能,採用薄膜過濾法的回收率爲40%,而採用培養基稀釋法的回收率爲30%,那麼應選擇薄膜過濾法進行該供試品的檢測。在此情況下,生產單位或研製單位應根據原輔料的微生物質量、生產工藝及產品特性進行產品的風險評估,以保證檢驗方法的可靠性,從而保證產品質量。
6.微生物限度檢查法中控制菌檢查法沒有規定進一步確證疑似致病菌的方法。若供試品檢出疑似致病菌,確證的方法應選擇已被認可的菌種鑑定方法,如細菌鑑定一般依據《伯傑氏細菌鑑定手冊》。
7.在藥品的生產、貯存、銷售及新藥標準制訂、進口藥品標準複覈、考察藥品質量、仲裁中,除在品種項下及製劑通則項下另有規定外,其微生物限度均以藥典的“藥品微生物限度標準”(2010年版藥典二部附錄Ⅺ J)爲依據。
8.對於《中國藥典》2010年版二部化學藥製劑通則項下有微生物限度要求的製劑,微生物限度爲必檢項目;對於只有原則性要求的製劑(如:丸劑、口服片劑、膠囊劑、顆粒劑),應對其被微生物污染的風險進行評估。在保證產品對患者安全的前提下,通過回顧性驗證或在線驗證積累的微生物污染數據表明每批均符合微生物限度標準的要求,那麼可不進行批批檢驗,但必須保證每批最終產品均符合微生物限度標準規定。上述固體制劑若因製劑本身及工藝的原因導致產品易受微生物污染,應在品種項下列出微生物限度檢查項及微生物限度標準,如生化類製劑。
9.用於手術、燒傷及嚴重創傷的局部給藥製劑應符合無菌檢查法要求。對用於創傷程度難以判斷的局部給藥製劑,若沒有證據證明藥品不存在安全性風險,那麼該藥品應符合無菌檢查法要求。
10.制定藥品的微生物限度標準時,除了依據“藥品微生物限度標準”(2010年版藥典二部附錄Ⅺ J)外,還應綜合考慮原料來源、性質、生產工藝條件、給藥途徑及微生物污染對患者的潛在危險等因素,提出合理安全的微生物限度標準,因此,必要時,特殊品種爲保證其療效、穩定性及避免對患者的潛在危害性,應制定更嚴格的微生物限度標準,並在品種項下規定,如吸入粉霧劑,從使用者的安全性考慮,應按無菌產品的要求進行控制。
17 附錄XIX Q 藥品微生物實驗室質量管理指導原則
藥品微生物實驗室質量管理[2]指導原則用於指導藥品微生物檢驗實驗室的質量控制。
藥品微生物的檢驗結果受很多因素的影響,如樣品中微生物可能分佈不均勻、微生物檢驗方法的誤差較大等。因此,在藥品微生物檢驗中,爲保證檢驗結果的可靠性,必須使用經驗證的檢測方法並嚴格按照藥品微生物實驗室質量管理指導原則要求[2]進行試驗。
藥品微生物實驗室質量管理指導原則[2]包括以下幾個方面:人員、培養基、試劑、菌種、環境、設備、樣品、檢驗方法、污染廢棄物處理、檢測結果質量保證和檢測過程質量控制、實驗記錄、結果的判斷、檢測報告、文件[2]等。
17.1 人員
從事藥品微生物試驗工作的人員應具備微生物學或相近專業知識的教育背景。
實驗人員應依據所在崗位和職責接受相應的培訓,在確認他們可以承擔某一試驗前,他們不能獨立從事該項微生物試驗。應保證所有人員在上崗前接受勝任工作所必需的設備操作、微生物檢驗技術等方面的培訓,如無菌操作、培養基製備、消毒、滅菌、注平板、菌落計數、菌種的轉種、傳代和保藏、微生物檢查方法及鑑定基本技術等,經考覈合格後方可上崗。
實驗人員應經過實驗室生物安全方面的培訓,保證自身安全,防止微生物在實驗室內部污染。
實驗室應制定所有級別實驗人員的繼續教育計劃,保證知識與技能不斷的更新。[2]
檢驗人員必須熟悉相關檢測方法、程序、檢測目的和結果評價。微生物實驗室的管理者其專業技能和經驗水平應與他們的職責範圍相符,如:管理技能、實驗室安全、試驗安排、預算、實驗研究、實驗結果的評估和數據偏差的調查、技術報告書寫等。
實驗室應通過參加內部質量控制、能力驗證或使用標準菌株等方法客觀評估檢驗人員的能力,必要時對其進行再培訓並重新評估。當使用一種非經常使用的方法或技術時,有必要在檢測前確認微生物檢測人員的操作技能。
所有人員的培訓、考覈內容和結果均應記錄歸檔。
17.2 培養基
培養基是微生物試驗的基礎,直接影響微生物試驗結果。適宜的培養基製備方法、貯藏條件和質量控制試驗是提供優質培養基的保證。
1.培養基的製備
微生物實驗室使用的[2]培養基可按處方配製,也可使用按處方生產的符合規定的脫水培養基。
在製備培養基時,應選擇質量符合要求的脫水培養基或單獨配方組分進行配製。脫水培養基應附有處方和使用說明,配製時應按使用說明上的要求操作以確保培養基的質量符合要求,不得使用結塊或顏色發生改變的脫水培養基。脫水培養基或單獨配方組分應在適當的條件下貯藏,如低溫、乾燥和避光,所有的容器應密封,尤其是盛放脫水培養基的容器。商品化的成品培養基除了應附有處方和使用說明外,還應註明有效期、貯藏條件、適用性檢查試驗的質控菌和用途。
爲保證培養基質量的穩定可靠,各脫水培養基或各配方組分應準確稱量,並要求有一定的精確度。配製培養基最常用的溶劑是純化水,特殊情況下,可能需要用去離子水和蒸餾水。應記錄各稱量物的重量和水的使用量。
配製培養基所用容器不得影響培養基質量,一般爲玻璃容器。[2]培養基配製所用容器和配套器具應潔淨,可用純化水沖洗玻璃器皿以消除清潔劑和外來物質的殘留。對熱敏感的培養基如糖發酵培養基其分裝容器一般應預先進行滅菌,以保證培養基的無菌性。
脫水培養基應完全溶解於水中,再行分裝與滅菌。配製時若需要加熱助溶,應注意不要過度加熱,以避免培養基顏色變深。如需要添加其他組分時,加入後應充分混勻。
應按照生產商提供或使用者驗證的參數進行培養基的滅菌。商品化的成品培養基必須附有所用滅菌方法的資料。培養基滅菌一般採用溼熱滅菌技術,特殊培養基可採用薄膜過濾除菌。
培養基若採用不適當的加熱和滅菌條件,有可能引起顏色變化、透明度降低、瓊脂凝固力或pH的改變。因此,培養基應採用驗證的滅菌程序滅菌,培養基滅菌方法和條件,應通過無菌性試驗和促生長試驗進行驗證。此外,對高壓滅菌器的蒸汽循環系統也要加以驗證,以保證在一定裝載方式下的正常熱分佈。溫度緩慢上升的高壓滅菌器可能導致培養基的過熱,過度滅菌可能會破壞絕大多數的細菌和真菌培養基促生長的質量。滅菌器中培養基的容積和裝載方式也將影響加熱的速度。因此,應根據滅菌培養基的特性,進行全面的滅菌程序驗證。
應確定每批培養基滅菌後的pH值(冷卻至室溫25℃測定)。若培養基處方中未列出pH值的範圍,除非經驗證表明培養基的pH值允許的變化範圍很寬,否則,pH值的範圍不能超過規定值±0.2。
製成平板或分裝於試管的培養基應進行下列檢查:容器和蓋子不得破裂,裝量應相同,儘量避免形成氣泡,固體培養基表面不得產生裂縫或漣漪,在冷藏溫度下不得形成結晶,不得污染微生物等。應檢查和記錄批數量、有效期及培養基的無菌檢查。
自配的培養基應標記名稱、批號、配製日期、配置人[2]等信息,並在已驗證的條件下貯藏。商品化的成品培養基標籤上應標有名稱、批號、生產日期、失效期及培養基的有關特性,生產商和使用者應根據培養基使用說明書上的要求進行貯藏,所採用的貯藏和運輸條件應使成品培養基最低限度的失去水分並提供機械保護。
培養基滅菌後若貯藏在高壓滅菌器中,質量可能會受影響,一般不提倡這種存放法。瓊脂培養基不得在0℃或0℃以下存放,因爲冷凍可能破壞凝膠特性。培養基應避光保存,若要長期保存,應置於密閉容器中以防止水分流失。瓊脂平板最好現配現用,如置冰箱保存,一般不超過1周,且應密閉包裝,若延長保存期限,保存期需經驗證確定。
固體培養基滅菌後的再融化只允許1次,以避免因過度受熱造成培養基質量下降或微生物污染。培養基的再融化一般採用水浴或流通蒸汽加熱。若採用其他溶解方法,應對其進行評估,確認該溶解方法不影響培養基質量。[2]融化的培養基應置於45~50℃的水浴中,不得超過8小時。傾注培養基時,應擦乾培養基容器外表面的水分,避免容器外壁的水滴進入培養基中造成污染。
使用過的培養基(包括失效的培養基)應按照國家污染廢物處理相關規定進行。
3.質量控制試驗
實驗室應對試驗用培養基建立質量控制程序,以確保所用培養基質量符合相關檢測的需要。
實驗室配製或商品化的成品培養基的質量依賴於其製備過程,採用不適宜方法製備的培養基將影響微生物的生長或復甦,從而影響試驗結果的可靠性。
所有配製好的培養基均應進行質量控制試驗。實驗室配製的培養基的常規監控項目是pH、適用性檢查試驗,定期的穩定性檢查以確定有效期。培養基在有效期內應依據適用性檢查試驗確定培養基質量是否符合要求。有效期的長短將取決於在一定存放條件下(包括容器特性及密封性)的培養基其組成成分的穩定性。
除藥典附錄另有規定外,在實驗室中,若採用已驗證的配製和滅菌程序製備培養基且過程受控,那麼同一批脫水培養基的適用性檢查試驗可只進行1次。如果培養基的製備過程未經驗證,那麼每一批培養基均要進行適用性檢查試驗,試驗的菌種可根據培養基的用途從相關附錄中進行選擇,也可增加從生產環境及產品中常見的污染菌株。
培養基的質量控制試驗若不符合規定,應尋找不合格的原因,以防止問題重複出現。任何不符合要求的培養基均不能使用。
用於環境監控的培養基須特別防護,最好要雙層包裝和終端滅菌,如果不能採用終端滅菌的培養基,那麼在使用前應進行100%的預培養以防止外來的污染物帶到環境中及避免出現假陽性結果。
17.3 試劑
微生物實驗室應有試劑接收、檢查和貯藏的程序,以確保所用試劑質量符合相關檢查要求。
實驗用的關鍵試劑,在開啓和貯藏過程中,應對每批試劑的適用性進行驗證。實驗室應對試劑進行管理控制,保存和記錄相關資料。
實驗室應標明所有試劑、試液及溶液的名稱、製備依據、適用性、濃度、效價、貯藏條件、製備日期、有效期及製備人。[2]
17.4 菌種
試驗過程中,生物樣本可能是最敏感的,因爲它們的活性和特性依賴於合適的試驗操作和貯藏條件。實驗室菌種的處理和保藏的程序應標準化,使盡可能減少菌種污染和生長特性的改變。按統一操作程序製備的菌株是微生物試驗結果一致性的重要保證。
藥品微生物檢驗用的試驗菌應來自認可的國內或國外菌種保藏機構的標準菌株,或使用與標準菌株所有相關特性等效的可溯源的[2]商業派生菌株。
標準菌株的復活或培養物的製備應按供應商提供的說明或按已驗證的方法進行。從國內或國外菌種保藏機構獲得的標準菌株經過復活並在適宜的培養基中生長後,即爲標準儲備菌株。標準儲備菌株應進行純度和特性確認。標準儲備菌株保存時,可將培養物等份懸浮於抗冷凍的培養基中,並分裝於小瓶中,建議採用低溫冷凍乾燥、液氮貯存、超低溫冷凍(低於- 30℃)等方法保存。低於-70℃或低溫冷凍乾燥方法可以延長菌種保存時間。標準儲備菌株可用於製備每月或每週1次轉種的工作菌株。冷凍菌種一旦解凍轉種製備工作菌株後,不得重新冷凍和再次使用。
工作菌株的傳代次數應嚴格控制,不得超過5代(從菌種保藏機構獲得的標準菌株爲第0代),以防止過度的傳代增加菌種變異的風險。1代是指將活的培養物接種到微生物生長的新鮮培養基中培養,任何亞培養的形式均被認爲是轉種或傳代1次。必要時,實驗室應對工作菌株的特性和純度進行確認。
工作菌株不可替代標準菌株,標準菌株的商業衍生物僅可用作工作菌株。標準菌株如果經過確認試驗證明已經老化、退化、變異、污染等或該菌株已無使用需要時,應及時滅菌銷燬。[2]
實驗室必須建立和保存其所有菌種的進出、收集、貯藏、確認試驗以及銷燬的記錄,應有菌種管理的程序文件(從標準菌株到工作菌株),該程序包括:標準菌種的申購記錄;從標準菌株到工作菌株操作及記錄;菌種必須定期轉種傳代,並做純度、特性等實驗室所需關鍵指標的確認,並記錄;每支菌種都應註明其名稱、標準號、接種日期、傳代數;菌種生長的培養基和培養條件;菌種保藏的位置和條件;其他需要的程序。
{
17.5 環境
微生物實驗室應具有進行微生物檢測所需的適宜、充分的設施條件。實驗環境應保證不影響檢驗結果的準確性。工作區域與辦公區域應分開。
微生物實驗室應專用,並與其他領域分開尤其是生產領域。
1.實驗室的佈局和運行
微生物實驗室的佈局與設計應充分考慮到試驗設備安裝、良好微生物實驗室操作規範和實驗室安全的要求。實驗室佈局設計的基本原則是既要最大可能防止微生物的污染,又要防止檢驗過程對環境和人員造成危害,同時還應考慮活動區域的合理規劃及區分,避免混亂和污染,以提高微生物實驗室操作的可靠性。
微生物實驗室的設計和建築材料應考慮其適用性,以利清潔、消毒、滅菌並減少污染的風險。潔淨或無菌室應配備獨立的空氣機組或空氣淨化系統,以滿足相應的檢驗要求,包括溫度和溼度的控制,壓力、照度和噪聲等都應符合工作要求。空氣過濾系統應定期維護和更換,並保存相關記錄。微生物實驗室應劃分成相應的潔淨區域和活菌操作區域,同時應根據試驗目的,在時間或空間上有效分隔不相容的試驗活動,將交叉污染的風險降低到最低。
活菌操作區應該配備生物安全櫃,以避免危害性的生物因子對實驗人員和實驗環境造成的危害。一般情況下,藥品微生物檢驗的實驗室應有符合無菌檢查法(2010年版藥典二部附錄Ⅺ H)和微生物限度檢查法(2010年版藥典二部附錄Ⅺ J)要求的、用於具有開展無菌檢查、微生物限度檢查、無菌採樣等檢測活動的、獨立設置的潔淨室(區)或隔離系統,併爲上述檢驗配備相應的陽性菌實驗室、培養室、試驗結果觀察區、培養基及實驗用具準備(包括滅菌)區、樣品接收和貯藏區、標準菌株貯藏區、污染物處理區和文檔處理區等輔助區域,同時,應對上述區域明確標識。
微生物實驗的各項工作應在專屬的區域進行,以降低交叉污染、假陽性結果和假陰性結果出現的風險。一些樣品若需要證明微生物的生長或進一步分析培養物的特性,如再培養、染色、微生物鑑定或其他確定試驗均應在實驗室的活菌操作區進行。任何出現微生物生長的培養物不得在實驗室無菌區域內打開。對染菌的樣品及培養物應有效隔離以減少假陽性結果的出現。病原微生物的分離鑑定工作應在二級生物安全實驗室進行。}[2]
{實驗室應對進出潔淨區域的人和物建立控制程序和標準操作規程,對可能影響檢驗結果的工作(如潔淨度驗證及監測、消毒、清潔維護等)能夠有效地控制、監測並記錄。微生物實驗室使用權限應限於經授權的工作人員,實驗人員應瞭解潔淨區域的正確進出的程序,包括更衣流程;該潔淨區域的預期用途、使用時的限制及限制原因;適當的潔淨級別。
2.環境監測
微生物實驗室應按相關國家標準制定完整的潔淨室(區)和隔離系統的驗證和環境監測標準操作規程,環境監測項目和監測頻率及對超標結果處理應有書面程序。監測項目應涵蓋到位,包括對空氣懸浮粒子、浮游菌、沉降菌、表面微生物及物理參數(溫度、相對溼度、換氣次數、氣流速度、壓差、噪聲等)的有效地控制和監測。
微生物實驗室應有制定清潔、消毒和衛生的標準操作規程,規程中應涉及環境監測結果。
實驗室在使用前和使用後應進行消毒,並定期監測消毒效果,要有足夠洗手和手消毒設施。應有對有害微生物發生污染的處理規程。
所用的消毒劑種類應滿足潔淨實驗室相關要求並定期更換。理想的消毒劑既能殺死廣泛的微生物、對人體無毒害、不會腐蝕或污染設備,又應有清潔劑的作用、性能穩定、作用快、殘留少、價格合理。所用消毒劑和清潔劑的微生物污染狀況應進行監測,並在規定的有效期內使用,A級和B級潔淨區應當使用無菌的或經無菌處理的消毒劑和清潔劑。}[2]
17.6 設備
微生物實驗室應配備與檢驗能力和工作量相適應的儀器設備,其類型、測量範圍和準確度等級應滿足檢驗所採用標準的要求,設備的安裝和佈局應便於操作,易於維護、清潔和校準,並保持清潔和良好的工作狀態。用於試驗的每臺儀器、設備應該有唯一標識。
儀器設備應有合格證書,[2]實驗室在儀器設備完成相應的檢定、校準、驗證、確認其性能,並形成相應的操作、維護和保養的標準操作規程後方可正式使用,儀器設備使用和日常監控要有記錄。
1.設備的維護
爲保證儀器設備處於良好工作狀態,應定期對其進行維護和性能驗證,並保存相關記錄。儀器設備若脫離實驗室或被檢修,恢復使用前應對其檢查或校準,以保證性能符合要求。[2]
微生物實驗室所用的儀器應根據日常使用的情況進行定期的校準,並記錄。校準的週期和校驗的內容根據儀器的類型和設備在實驗室產生的數據的重要性的不同而不同。重要的儀器設備,如培養箱、冰箱等,應由專人負責,保證其運行狀態正常和受控,同時應有相應的備用設備以保證試驗菌株和微生物培養的連續性,特殊設備如高壓滅菌器、隔離器、生物安全櫃等實驗人員應經培訓後持證上崗。[2]對於培養箱、冰箱、高壓滅菌鍋等影響實驗準確性的關鍵設備應在其運行過程中對關鍵參數(如溫度、壓力)進行連續觀測和記錄,有條件的情況下儘量使用自動記錄裝置。如果發生偏差,應評估對以前的檢測結果造成的影響並採取必要的糾正措施。對於一些容易污染微生物的儀器設備如水浴鍋、培養箱、冰箱和生物安全櫃等應定期進行清潔和消毒。
對試驗需用的無菌器具應實施正確的清洗、滅菌措施,並形成相應的標準操作規程,無菌器具應有明確標識並與非無菌器具加以區別。
{實驗室的某些設備(例如培養箱、高壓滅菌器和玻璃器皿等)應專用,除非有特定預防措施,以防止交叉污染。
微生物實驗室所用的儀器應根據日常使用的情況進行定期的校準,並記錄。校準的週期和校驗的內容根據儀器的類型和設備在實驗室產生的數據重要性不同而不同。儀器上應有標籤說明校準日期、維修日期和重新校準日期。
溫度測量裝置
溫度不但對實驗結果有直接的影響,而且還對儀器設備的正常運轉和正確操作起關鍵因素。相關的溫度測量裝置如培養箱和高壓滅菌器中的溫度計、熱電耦和鉑電阻溫度計,應具有可靠的質量並進行校準以確保所需的精確度,溫度設備的校準應遵循國家或國際標準。
溫度測量裝置可以用來監控冰箱、超低溫冰箱、培養箱、水浴鍋等設備的溫度,應在使用前驗證此類裝置的性能。
稱量設備
天平和標準砝碼應定期進行校準,天平使用過程應採用標準砝碼進行校準。每次使用完後應及時清潔,必要時用非腐蝕消毒劑進行消毒。
容量測定設備
微生物實驗室對容量測定設備如自動分配儀、移液槍、移液管等應進行檢定,以確保儀器準確度。對於已經校準或檢定證明符合使用要求的玻璃器具可以不進行檢定。標有各種使用體積的儀器需要對使用時的體積進行精密度的檢查,並且還要測定其重現性。
對於一次性使用的容量設備,實驗室應該從公認的和具有相關質量保證系統的公司購買。對儀器適用性進行初次驗證後,要對其精密度隨時進行檢查。必要時應該對每批定容設備進行適用性檢查。
應由有資質的人員進行生物安全櫃、層流超淨工作臺及高效過濾器的安裝與更換,要按照確認的方法進行現場生物和物理的檢測,並定期進行再驗證。
實驗室生物安全櫃和層流超淨工作臺的通風應符合微生物風險級別及符合安全要求。應定期對生物安全櫃、層流超淨工作臺進行監測以確保其性能符合相關要求。實驗室應保存檢查記錄和性能測試結果。
其他設備
懸浮粒子計數器、浮游菌採樣器應定期進行校準;pH計、傳導計和其他類似儀器的性能應定期或在每次使用前確認;若溼度對實驗結果有影響,溼度計應按國家或國際標準進行校準;當所測定的時間對檢測結果有影響時,應使用校準過的計時儀或定時器;使用離心機時,應評估離心機每分鐘的轉數,若離心是關鍵因素,離心機應該進行校準。}[2]
{
17.7 樣品
1.樣品採集
試驗樣品的採集,應遵循隨機抽樣的原則,並在受控條件下進行抽樣,如有可能,抽樣應在具有無菌條件的特定抽樣區域中進行。抽樣時,須採用無菌操作技術進行取樣,防止樣品受到微生物的污染而導致假陽性的結果。抽樣的任何消毒過程(如抽樣點的消毒)不能影響樣品中微生物的檢出。
抽樣的容器應貼有唯一性的標識,註明樣品名稱、批號、抽樣日期、採樣容器、抽樣人等。抽樣應由經過培訓的人員使用無菌設備在無菌條件下進行無菌操作。抽樣環境狀況應監測並記錄,同時還需記錄採樣時間。}[2]
{
2.樣品儲存和運輸
待檢樣品應在合適的條件下貯藏並保證其完整性,儘量減少污染的微生物發生變化。樣品在運輸過程中,應保持原有(規定)的儲存條件或採取必要的措施(如冷藏或冷凍)。應明確規定和記錄樣品的貯藏和運輸條件。
實驗室在收到樣品後應根據有關規定儘快對樣品進行檢查,並記錄被檢樣品所有相關信息,如:接收日期及時間、接收
時樣品的狀況、採樣操作的特徵(包括採樣日期和採樣條件等)、貯藏條件。
如果樣品存在數量不足、包裝破損、標籤缺失、溫度不適等,實驗室應在決定是否檢測或拒絕接受樣品之前與相關人員溝通。樣品的包裝和標籤有可能被嚴重污染,因此搬運和儲存樣品時應小心以避免污染的擴散,容器外部的消毒應不影響樣品的完整性。樣品的任何狀況在檢驗報告中應有說明。
選擇具有代表性的樣品,根據有關的國家或國際標準,或者使用經驗證的實驗方法,儘快進行檢驗。
實驗室應按照書面管理程序對樣品進行保留和處置。如果實驗用的是已知被污染的樣品,應該在丟棄前進行滅菌。檢驗方法
藥品微生物檢驗時,應根據檢驗目的選擇適宜的方法進行樣品檢驗。
藥典方法或標準中規定的方法是經過驗證的,當進行樣品檢驗時,應進行方法適用性確認。
如果檢驗方法不是藥典或標準中規定的方法,使用前應進行替代方法的驗證,確認其應用效果優於或等同於藥典方法。替代方法的驗證按藥品微生物檢驗替代方法驗證指導原則(2010年版藥典二部附錄XIX O)進行。
實驗室對所用商業檢測系統如試劑盒等應保留確認數據,這些確認數據可由製造者提供或由第三方機構評估,必要時,實驗室應對商業檢測系統進行確認。}[2]
{
17.8 污染廢棄物處理
實驗室應有妥善處理廢棄樣品、過期(或失效)培養基和有害廢棄物的設施和制度,旨在減少檢查環境和材料的污染。污染廢棄物的最終處理必須符合國家環境和健康安全規定。實驗室還應針對類似於帶菌培養物溢出的意外事件制定處理規程。如:活的培養物灑出必須就地處理,不得使培養物污染擴散。}[2]
{
1.內部質量控制
爲保證實驗室在每個工作日檢測結果的連貫性和與檢測標準的一致性,實驗室應制定對所承擔的工作進行連續評估的程序。
實驗室應定期對實驗環境的潔淨度、培養基的適用性、滅菌方法、菌株純度和活性(包括性能)、試劑的質量等進行監控並詳細記錄。
實驗室應定期對檢測人員進行技術考覈。可以通過加標試樣的使用、平行實驗和參加能力驗證等方法使每個檢測人員所檢測項目的可變性處於控制之下,以保證檢驗結果的一致性。
2.外部質量評估
實驗室應參加與檢測範圍相關的國家能力驗證或實驗室之間的比對實驗來評估檢測水平,通過參加外部質量評估來評定檢測結果的偏差。}[2]
17.9 實驗記錄
實驗結果的可靠性依賴於試驗嚴格按照標準操作規程進行,而標準操作規程應指出如何進行正確的試驗操作。實驗記錄應包含所有關鍵的實驗細節,以便確認數據的完整性。實驗室原始記錄至少應包括以下內容:實驗日期、檢品名稱、實驗人員姓名、標準操作規程編號或方法、實驗結果、偏差(存在時)、實驗參數(所使用的設備、菌種、培養基和批號以及培養溫度等)、主管/複覈人簽名。
實驗記錄上還應顯示出檢驗標準的選擇,如果使用的是藥典標準,必須保證是現行有效的標準。
試驗所用的每一個關鍵的實驗設備均應有記錄,設備日誌或表格應設計合理,[2]以滿足試驗記錄的追蹤性,設備溫度(水浴、培養箱、滅菌器)必須記錄,且具有追溯性。
實驗記錄寫錯時,用單線劃掉並簽字。原來的數據不能抹去或被覆蓋。
所有實驗室記錄應以文件形式保存並防止意外遺失,正規的記錄應存放在特定的地方並有登記。
{
17.10 結果的判斷和檢測報告
由於微生物試驗的特殊性,在實驗結果分析時,對結果應進行充分和全面的評價。所有影響結果觀察的微生物條件和因素應完全考慮,包括與規定的限度或標準有很大偏差的結果;微生物在原料、輔料或試驗環境中存活的可能性;及微生物的生長特性等。特別要了解}[2]實驗結果與標準的差別
是否有統計學意義。
若發現實驗結果不符合藥典各品種項下要求或另外建立的質量標準,應進行原因調查。引起微生物污染結果不符合標準的原因主要有兩個:試驗操作錯誤或產生無效結果的試驗環境條件;產品本身的微生物污染總數超過規定的限度或檢出控制菌。
異常結果出現時,應進行偏差調查。偏差調查時[2]應考慮實驗室環境、抽樣區的防護條件、樣品在該檢驗條件下以往檢驗的情況、樣品本身具有使微生物存活或繁殖的特性等情況。此外,回顧試驗過程,也可評價該實驗結果的可靠性及實驗過程是否恰當。如果試驗操作被確認是引起實驗結果不符合的原因,那麼應制定改錯方案以解決問題,按照正確的操作方案進行實驗,在這種情況下,對試驗過程及試驗操作應特別認真地進行監控。
如果依據分析調查結果發現試驗有錯誤而判實驗結果無效,那麼這種情況必須記錄。實驗室也必須認可複試程序,如果需要,可按相關規定重新抽樣,但抽樣方法不能影響不符合規定結果的分析調查。
{微生物實驗室檢測報告應該符合檢測方法的要求。實驗室應準確、清晰、明確和客觀地報告每一項或每一份檢測的結果。
17.11 文件
文件應當充分表明試驗是在實驗室裏按可控的檢查法進行的,一般包括以下方面:人員培訓與資格確認;設備驗收、驗證、檢定(或校準期間覈查)和維修;設備使用中的運行狀態(設備的關鍵參數);培養基製備、貯藏和質量控制;菌種管理;檢驗規程中的關鍵步驟;數據記錄與結果計算的確認;質量責任人對試驗報告的評估;數據偏離的調查。
18 附錄XIX R 國家藥品標準物質製備指導原則
(附錄XIX R 國家藥品標準物質製備指導原則 由《中華人民共和國藥典》(2010年版 第二增補本)新增)
本指導原則用於規範和指導國家藥品標準物質的製備,保證國家藥品標準的執行。
18.1 一、國家藥品標準物質品種的確定
18.2 二、候選國家藥品標準物質原料的選擇
1.原料的選擇應滿足適用性、代表性及可獲得性的原則。
2.原料的性質應符合使用要求。
3.原料的均勻性、穩定性及相應特性量值範圍應適合該標準物質的用途。
18.3 三、候選國家藥品標準物質的製備
1.根據候選藥品標準物質的理化性質,選擇合理的製備方法和工藝流程,防止相應特性量值的變化,並避免被污染。
2.對不易均勻的候選藥品標準物質,在製備過程中除採取必要的均勻措施外,還應進行均勻性初檢。
3.對相應特性量值不穩定的候選藥品標準物質,在製備過程中應考察影響穩定性的因素,採取必要的措施保證其穩定性,並選擇合適的儲存條件。
4.當候選藥品標準物質製備量大時,爲便於保存可採取分級分裝。
5.候選藥品標準物質供應者須具備良好的實驗條件和能力,並應提供以下資料。
(1)試驗方法、量值、試驗重複次數、必要的波譜及色譜等資料;
(2)符合穩定性要求的儲存條件(溫度、溼度和光照等);
(3)候選藥品標準物質引溼性研究結果及說明;
(4)加速穩定性研究結果;
(5)有關物質的鑑別及百分比,國家藥品標準中主組分的相對響應因子等具體資料;
18.4 四、候選國家藥品標準物質的標定
候選藥品標準物質按以下要求進行標定,必要時應與國際標準物質進行比對。
18.4.1 1.化學結構或組分的確證
(1)驗證已知結構的化合物需要提供必要的理化參數及波譜數據,並提供相關文獻及對比數據。如無文獻記載,應提供完整的結構解析過程。
(2)對於不能用現代理化方法確定結構的藥品標準物質,應選用適當的方法對其組分進行確證。
18.4.2 2.理化性質檢查
應根據藥品標準物質的特性和具體情況確定理化性質檢驗項目,如性狀、熔點、比旋度、晶型以及乾燥失重、引溼性等。
18.4.3 3.純度及有關物質檢查
應根據藥品標準物質的使用要求確定純度及有關物質的檢查項,如反應中間體、副產物及相關雜質等。
18.4.4 4.均勻性檢驗
凡成批製備並分裝成最小包裝單元的候選藥品標準物質,必須進行均勻性檢驗。對於分級分裝的候選藥品標準物質,凡由大包裝分裝成最小包裝單元時,均應進行均勻性檢驗。
18.4.5 5.定值
符合上述要求後,方可進行定值。
定值的測量方法應經方法學考察證明準確可靠。應先研究測量方法、測量過程和樣品處理過程所固有的系統誤差和隨機誤差,如溶解、分離等過程中被測樣品的污染和損失;對測量儀器要定期進行校準,選用具有可溯源的基準物;要有可行的質量保證體系,以保證測量結果的溯源性。
18.4.5.1 (1)定值原則
在測定一個候選化學標準品/對照品含量時,水分、有機溶劑、無機雜質和有機成分測定結果的總和應爲100%。
18.4.5.2 (2)選用下列方式對候選藥品標準物質定值
①採用高準確度的絕對或權威測量方法定值測量時,要求兩個以上分析者在不同的實驗裝置上獨立地進行操作。
研究不同原理的測量方法的精密度,對方法的系統誤差進行估計,採取必要的手段對方法的準確度進行驗證。
③多個實驗室協作定值
參加協作標定的實驗室應具有候選藥品標準物質定值的必備條件及相關實驗室資質。每個實驗室應採用規定的測量方法。協作實驗室的數目或獨立定值組數應符合統計學的要求。
18.5 五、候選國家藥品標準物質的穩定性考察
1.候選藥品標準物質應在規定的儲存或使用條件下,定期進行相應特性量值的穩定性考察。
2.穩定性考察的時間間隔可以依據先密後疏的原則。在考察期間內應有多個時間間隔的監測數據。
(1)當候選藥品標準物質有多個特性量值時,應選擇易變的和有代表性的特性量值進行穩定性考察;
(2)選擇不低於定值方法精密度和具有足夠靈敏度的測量方法進行穩定性考察;
19 附錄XIX S 藥用輔料功能性指標研究指導原則
(附錄XIX S 藥用輔料功能性指標研究指導原則 由《中華人民共和國藥典》(2010年版 第三增補本)新增)
藥用輔料係指生產藥品和調配處方時使用的賦形劑和附加劑,是除活性成分以外,在安全性方面已進行了合理的評估,且包含在藥物製劑中的物質。藥用輔料按用途可以分爲多個類別(見附錄Ⅱ 藥用輔料),爲保證藥用輔料在製劑中發揮其賦形作用和保證質量的作用,在藥用輔料的正文中設置適宜的功能性指標(functionality-related characteristics,FRCs)十分必要。功能性指標的設置是針對特定用途的,同一輔料按功能性指標不同可以分爲不同的規格,使用者可根據用途選擇適宜規格的藥用輔料以保證製劑的質量。
本指導原則將按藥用輔料的用途介紹常用的功能性指標研究和建立方法。藥用輔料功能性指標主要針對一般的化學手段難以評價功能性的藥用輔料,如稀釋劑等十二大類;對於純化合物或功能性可以通過相應的化學手段評價的輔料,如pH調節劑、滲透壓調節劑、抑菌劑、螯合劑、絡合劑、矯味劑、着色劑、增塑劑、抗氧劑、拋射劑等,不在本指導原則中列舉其功能性評價方法。
19.1 一、稀釋劑
稀釋劑也稱填充劑,指製劑中用來增加體積或重量的成分。常用的稀釋劑包括澱粉、蔗糖、乳糖、預膠化澱粉、微晶纖維素、無機鹽類和糖醇類等。在藥物劑型中稀釋劑通常佔有很大比例,其作用不僅保證一定的體積大小,而且減少主藥成分的劑量偏差,改善藥物的壓縮成型性。稀釋劑類型和用量的選擇通常取決於它的物理化學性質,特別是功能性指標。稀釋劑可以影響製劑的成型性和製劑性能(如粉末流動性、溼法顆粒或幹法顆粒成型性、含量均一性、崩解性、溶出度、片劑外觀、片劑硬度和脆碎度、物理和化學穩定性等)。一些稀釋劑(如微晶纖維素)常被用作幹黏合劑,因爲它們在最終壓片的時候能賦予片劑很高的強度。
稀釋劑功能性指標包括:(1)粒徑和粒徑分佈(2010年版藥典二部附錄ⅨE);(2)粒子形態(2010年版藥典二部附錄Ⅸ E);(3)松密度/振實密度/真密度;(4)比表面積;(5)結晶性(2010年版藥典二部附錄Ⅸ D和2010年版藥典二部附錄Ⅸ F);(6)水分(2010年版藥典二部附錄Ⅷ L和2010年版藥典二部附錄Ⅷ M);(7)流動性;(8)溶解度;(9)壓縮性;(10)吸溼性(2010年版藥典二部附錄XIX J)等。
19.2 二、黏合劑
黏合劑是指一類使無黏性或黏性不足的物料粉末聚集成顆粒,或壓縮成型的具黏性的固體粉末或溶液。黏合劑在制粒溶液中溶解或分散,有些黏合劑爲乾粉。隨着制粒溶液的揮發,黏合劑使顆粒的各項性質(如粒度大小及其分佈、形態、含量均一性等)符合要求。溼法制粒通過改善顆粒一種或多種性質,如流動性、操作性、強度、抗分離性、含塵量、外觀、溶解度、壓縮性或者藥物釋放,使得顆粒的進一步加工更爲容易。
黏合劑可以被分爲:(1)天然高分子材料;(2)合成聚合物;或者(3)糖類。聚合物的化學屬性,包括結構、單體性質和聚合順序、功能基團、聚合度、取代度和交聯度將會影響制粒過程中的相互作用。同一聚合物由於來源或合成方法的不同,它們的性質可能顯示出較大的差異。常用黏合劑包括澱粉漿、纖維素衍生物、聚維酮、明膠和其他一些黏合劑。黏合劑通過改變微粒內部的黏附力生成了溼顆粒(聚集物)。它們可能還會改變界面性質、黏度或其他性質。在乾燥過程中,它們可能產生固體橋,賦予幹顆粒一定的機械強度。
黏合劑的功能性指標包括:(1)表面張力;(2)粒徑、粒徑分佈(檢查法見2010年版藥典二部附錄Ⅸ E);(3)溶解度;(4)黏度(檢查法見附錄ⅥG);(5)堆密度和振實密度;(6)比表面積等。
19.3 三、崩解劑
崩解劑是加入到處方中促使製劑迅速崩解成小單元並使藥物更快溶解的成分。當崩解劑接觸水分、胃液或腸液時,它們通過吸收液體膨脹溶解或形成凝膠,引起製劑結構的破壞和崩解,促進藥物的溶出。不同崩解劑發揮作用的機制主要有四種:膨脹、變形、毛細管作用和排斥作用。在片劑處方中,崩解劑的功能最好能具兩種以上。崩解劑的功能性取決於多個因素,如它的化學特性、粒徑及分佈以及粒子形態,此外還受一些重要的片劑因素的影響,如硬度和孔隙率。
崩解劑包括天然的、合成的或化學改造的天然聚合物。常用崩解劑包括:幹澱粉、羧甲基澱粉鈉、低取代羥丙基纖維素、交聯羧甲基纖維素鈉、交聯聚維酮、泡騰崩解劑等。崩解劑可爲非解離型或爲陰離子型。非解離態聚合物主要是多糖,如澱粉、纖維素、支鏈澱粉或交聯聚維酮。陰離子聚合物主要是化學改性纖維素的產物等。離子聚合物應該考慮其化學性質。胃腸道pH的改變或者與離子型原料藥(APIs)形成複合物都將會影響崩解性能。
與崩解劑功能性相關的性質包括:(1)粒徑及其分佈(檢查法見2010年版藥典二部附錄Ⅸ E);(2)水吸收速率;(3)膨脹率或膨脹指數;(4)粉體流動性;(5)水分;(6)泡騰量等。
19.4 四、潤滑劑
潤滑劑的作用爲減小顆粒間、顆粒和固體制劑製造設備如片劑衝頭和沖模的金屬接觸面之間的摩擦力。
潤滑劑可以分爲界面潤滑劑、流體薄膜潤滑劑和液體潤滑劑。界面潤滑劑爲兩親性的長鏈脂肪酸鹽(如硬脂酸鎂)或脂肪酸酯(如硬脂酰醇富馬酸鈉),可附着於固體表面(顆粒和機器零件),減小顆粒間或顆粒、金屬間摩擦力而產生作用。表面附着受底物表面的性質影響,爲了最佳附着效果,界面潤滑劑顆粒往往爲小的片狀晶體;流體薄膜潤滑劑是固體脂肪(如氫化植物油,1型),甘油酯(甘油二十二烷酸酯和二硬脂酸甘油酯),或者脂肪酸(如硬脂酸),在壓力作用下會熔化並在顆粒和壓片機的衝頭周圍形成薄膜,這將有利於減小摩擦力。在壓力移除後流體薄膜潤滑劑重新固化;液體潤滑劑是在壓緊之前可以被顆粒吸收,而壓力下可自顆粒中釋放的液體物質,也可用於減小製造設備的金屬間摩擦力。
常用潤滑劑包括:硬脂酸鎂、微粉硅膠、滑石粉、氫化植物油、聚乙二醇類、月桂醇硫酸鈉。
潤滑劑的主要功能性指標包括:(1)粒徑及其分佈(檢查法見2010年版藥典二部附錄Ⅸ E);(2)表面積;(3)水分(檢查法見2010年版藥典二部附錄Ⅷ L和2010年版藥典二部附錄Ⅷ M);(4)多晶型(檢查法見2010年版藥典二部附錄Ⅸ D和2010年版藥典二部附錄Ⅸ F);(5)純度(例如硬脂酸鹽:棕櫚酸鹽比率);(6)熔點或熔程等;(7)粉體流動性。
19.5 五、助流劑和抗結塊劑
助流劑和抗結塊劑的作用是提高粉末流速和減少粉末聚集結塊。助流劑和抗結塊劑通常是無機物質細粉。它們不溶於水但是不疏水。其中有些物質是複雜的水合物。常用助流劑和抗結塊劑包括:滑石粉、微粉硅膠等無機物質細粉。
助流劑可吸附在較大顆粒的表面,減小顆粒間黏着力和內聚力,使顆粒流動性好。此外,助流劑可分散於大顆粒之間,減小摩擦力。抗結塊劑可吸收水分以阻止結塊現象中顆粒橋的形成。
助流劑和抗結塊劑的功能性指標包括:(1)粒徑及其分佈(檢查法見2010年版藥典二部附錄Ⅸ E);(2)表面積;(3)吸收率等。
19.6 六、空心膠囊
膠囊作爲藥物粉末和液體的載體可以保證劑量的準確和運輸的便利。空心膠囊應與內容物相容。空心膠囊通常包括兩個部分(即膠囊帽和膠囊體),都是圓柱狀,其中稍長的稱爲膠囊體,另一個稱爲膠囊帽。膠囊帽和膠囊體緊密結合以閉合膠囊。軟膠囊是由沿軸縫合或無縫合線的單片構成。
根據原料不同空心膠囊可分爲明膠空心膠囊和其他膠囊。明膠空心膠囊由源於豬、牛、或魚的明膠製備;其他類型膠囊由非動物源的纖維素、多糖等製備。空心膠囊也含其他添加劑如增塑劑、着色劑、遮光劑和抑菌劑。應儘量少用或不用抑菌劑,空心膠囊所用添加劑的種類和用量應符合國家食用或藥用相關標準和要求。
空心膠囊可裝填固體、半固體和液體制劑。傳統的空心膠囊應在37℃生物液體如胃腸液裏迅速溶化或崩解。空心膠囊中可以引入腸溶材料和控釋的聚合物,控制膠囊內容物的釋放。
水分隨着膠囊類型而變化,水分對膠囊脆度有顯著的影響。平衡水分對劑型穩定性有關鍵作用,因爲水分子可在膠囊內容物和膠囊殼之間遷移。透氣性是假重要的一個指標,因爲羥丙甲纖維素膠囊有開放結構,因而通常其膠囊透氣性比一般膠囊更大。明膠膠囊貯藏於較高的溫度和溼度(如40℃/75% RH)下可產生交聯,而羥丙甲纖維素膠囊不會產生交聯。粉末內容物裏的醛類物質因爲能夠使明膠交聯而延長崩解時間。明膠膠囊在0.5%鹽酸條件和36~38℃但不低於30℃的條件下應該能夠在15分鐘內崩解。羥丙甲纖維素膠囊在30℃以下也能崩解。
膠囊殼的功能性指標包括:(1)水分(見2010年版藥典二部附錄Ⅷ L和2010年版藥典二部附錄Ⅷ M);(2)透氣性;(3)崩解性(見2010年版藥典二部附錄Ⅹ B和2010年版藥典二部附錄Ⅹ C);(4)脆碎度;(5)韌性;(6)凍力強度;(7)鬆緊度等。
19.7 七、包衣材料
包衣可以掩蓋藥物異味、改善外觀、保護活性成分、調節藥物釋放。包衣材料包括天然、半合成和合成材料。它們可能是粉末或者膠體分散體系(膠乳或僞膠乳),通常製成溶液或者水相及非水相體系的分散液。蠟類和脂類在其熔化狀態時可直接用於包衣,而不使用任何溶劑。
包衣材料的功能性研究應針對:(1)溶解性,如腸溶包衣材料不溶於酸性介質而溶於中性介質;(2)成膜性;(4)黏度;(5)取代基及取代度;(6)抗拉強度;(7)透氣性;(8)粒度等。
19.8 八、潤溼劑和(或)增溶劑
增溶劑包含很多種不同的化學結構和等級。典型的增溶劑爲陰離子型非解離型表面活性劑,在水中自發形成的膠柬形態和結構,起到增溶作用。增溶機理常常與難溶性藥物和增溶劑自組裝體(如膠束)形成的內核間的相互作用力有關。還有一些類型的增溶劑利用與疏水性分子相互作用的聚合物鏈的變化,將難溶性藥物溶入聚合物鏈中從而增加藥物的溶解度。
增溶劑包括固態、液態或蠟質材料。它們的化學結構決定其物理特性。然而增溶劑的物理特性和功效取決於表面活性特性和親水親油平衡值(HLB)(檢查法見2010年版藥典二部附錄Ⅶ H)。HLB值低的可以用作乳化劑,而HLB值高的可以作爲增溶劑。例如,十二烷基硫酸鈉(HLB值爲40)是親水性的,易溶於水,一旦在水中分散,即自發形成膠束。增溶劑特殊的親水和親油特性可以由其臨界膠束濃度(CMC)來表徵。
增溶劑往往可以作爲潤溼劑。常用潤溼劑和(或)增溶劑包括肥皂類、硫酸化物、磺酸化物、吐溫類、賣澤類、聚氧乙烯脂肪醇醚類以及聚維酮等。
與潤溼劑/增溶劑有關的性能指標包括:(1) HLB值;(2)黏度;(3)組成,檢查法可參考2010年版藥典二部附錄Ⅳ、2010年版藥典二部附錄Ⅵ A、2010年版藥典二部附錄Ⅵ G、附錄ⅥH、錄Ⅵ H、2010年版藥典二部附錄Ⅶ H、2010年版藥典二部附錄Ⅷ Q和2010年版藥典二部附錄Ⅸ E等;(4)臨界膠束濃度等;(5)表面張力。
19.9 九、栓劑基質
栓劑基質爲製造直腸栓劑和陰道栓劑的基質。常用栓劑基質包括:油脂性基質,如可可豆脂、半合成椰油酯、半合成或全合成脂肪酸甘油酯等;水溶性基質,如甘油明膠、聚乙二醇、泊洛沙姆等。
栓劑應在略低於體溫(37℃)下融化或溶解而釋放藥物,如果藥物溶於基質中,其釋放機制爲溶蝕或擴散分配機制,如果藥物懸浮於基質中則通過溶蝕和溶出機制釋放藥物。高熔點脂肪栓劑基質在體溫條件下應融化。水溶性基質應能夠溶解或分散於水性介質中,藥物釋放機制是溶蝕和溶出機制。栓劑基質最重要的物理性質便是它的融程。一般來說,栓劑基質的融程在27~45℃。然而,單一栓劑基質的融程較窄,通常在2~3℃之間。基質融程的選擇應考慮其他處方成分對最終產品融程的影響。
高熔點親脂性栓劑基質是半合成的長鏈脂肪酸甘油三酯的混合物,包括單甘油酯、雙甘油酯,也可能存在乙氧化脂肪酸。根據基質的融程、羥值、酸值、碘值、凝固點和皂化值,可將基質分爲不同的級別。
親水性栓劑基質通常是親水性半固體材料的混合物,在室溫條件下爲固體,而當用於病人時,藥物會通過基質的熔融、溶蝕和溶出機制而釋放出來。相對於高熔點栓劑基質,親水性栓劑基質有更多羥基和其他親水性基團。聚乙二醇爲一種親水性基質,具有合適的融化和溶解行爲。
因此,栓劑基質的功能性指標可參考2010年版藥典二部附錄Ⅰ D、2010年版藥典二部附錄Ⅵ C和2010年版藥典二部附錄Ⅵ D、2010年版藥典二部附錄Ⅶ H等。
19.10 十、助懸劑和(或)增稠劑
在藥物製劑中,助懸劑和(或)增稠劑用於穩定分散系統(例如混懸劑或乳劑),其機制爲減少溶質或顆粒運動的速率,或降低液體制劑的流動性。
助懸劑、增稠劑穩定分散體系或增稠效應有多種機制。常見的是大分子鏈或細黏土束縛溶劑導致黏度增加和層流中斷。其餘包括製劑中的輔料分子或顆粒形成三維結構的凝膠,和大分子或礦物質吸附於分散顆粒或液滴表面產生的立體作用。每種機制(黏度增加,凝膠形成或立體穩定性)是輔料流變學特性的體現,由於輔料的分子量大和粒徑較大,其流變學的性質爲非牛頓流體。此類輔料的分散體表現出一定的黏彈性。
助懸劑或增稠劑可以是低分子也可以是大分子或礦物質。低分子助懸劑或增稠劑如甘油、糖漿。大分子助懸劑或增稠劑包括(a)親水性的碳水化合物高分子[阿拉伯膠、瓊脂、海藻酸、羧甲基纖維素、角叉(菜)膠、糊精、結冷膠、瓜爾豆膠、羥乙基纖維素、羥丙基纖維素、羥丙甲纖維素、麥芽糖糊精、甲基纖維素、果膠、丙二醇海藻酸、海藻酸鈉、澱粉、西黃芪膠和黃原膠樹膠]和(b)非碳水化合物親水性大分子,包括明膠、聚維酮、卡波姆、聚氧乙烯和聚乙烯醇。礦物質助懸劑或增稠劑包括硅鎂土、皁土(斑脫土)、硅酸鎂鋁、二氧化硅等。單硬脂酸鋁,按功能分類既非大分子也非礦物質類助懸劑或增稠劑。它主要包含不同組分比例的單硬脂酸鋁和單棕櫚酸鋁。
助懸劑和增稠劑的功能性指標爲黏度(2010年版藥典二部附錄Ⅵ G)等。
19.11 十一、軟膏基質
軟膏是黏稠的用於體表不同部位的半固體外用製劑。軟膏基質是其主要組成成分並決定其物理性質。軟膏基質可作爲藥物的外用載體並可作爲潤溼劑和皮膚保護劑。
軟膏基質分爲(a)油性基質:不溶於水,無水、不吸收水,難以用水去除(如凡士林);(b)吸收性軟膏基質:無水,但能夠吸收一定量的水,不溶於水而且不易用水去除(如羊毛脂);(c)乳劑型基質:通常是水包油或油包水型,其中含水,能夠吸收水分,在水中也無法溶解(如乳膏);(d)水溶性軟膏基質:本身無水,可以吸水,能溶於水,可用水去除(如聚乙二醇)。被選擇的軟膏基質應惰性、化學穩定。
黏度和熔程是乳膏基質的重要功能性指標,可參見2010年版藥典二部附錄Ⅵ G和2010年版藥典二部附錄Ⅵ C。