1 概述
急性髓細胞白血病(acute myelocytic leukemia,AML) 或急性非淋巴細胞白血病(ANLL)包括所有非淋巴細胞來源的急性白血病。它是多能幹細胞或已輕度分化的前體細胞核型發生突變所形成的一類疾病,是造血系統的克隆性惡性疾病。人羣接受大劑量放射線或長期接觸苯可增加這類疾病的發病率。急性髓細胞白血病是一個具有高度異質性的疾病羣,它可以由正常髓系細胞分化發育過程中不同階段的造血祖細胞惡性變轉化而來,起源於不同階段祖細胞的AML具有不同的生物學特徵。根據細胞形態學和組織化學特徵將AML分爲不同的類型,如FAB分型中M0~M7型。近年來,隨着免疫學、細胞遺傳學和分子生物學等技術的應用,對AML腫瘤細胞的生物學特性有了更深入的認識和了解,爲AML的精確分型、診斷、預後和最佳治療方法的選擇奠定了基礎。目前,對於AML的治療,除急性早幼粒細胞白血病外,仍以聯合化療爲主。AML患者總的完全緩解率僅50%~70%,長期無病生存率爲25%~30%。
4 別名
ANLL;急非淋;急粒;急性非淋巴細胞白血病;急性髓細胞性白血病
7 流行病學
流行病學調查顯示,環境、職業及遺傳因素與急性髓細胞白血病的發病關係密切。發達國家的發病率高於發展中國家,西方國家高於東方國家。世界各地年發病率爲2.25/10萬人口,隨年齡增加而發病率增高,30歲以下爲1/10萬、75歲以上則高達17/10萬。因此,AML實際是一種中、老年病,佔成人急性白血病的80%~90%,但僅佔兒童急性白血病的15%~20%。男性發病高於女性。1986~1988年國內對22個省市自治區進行白血病流行病學調查,總人口達6000餘萬,AML的年發病率爲1.6/10萬人口,佔各型白血病的58.9%。它隨年齡增長髮病率上升,50歲開始明顯上升,60~69歲達高峯,男性也明顯多於女性。
8 急性髓細胞白血病的病因
8.1 化學物質
長期密切接觸有機溶劑者,發生急性髓細胞白血病的危險升高,中國一組流行病學調查顯示,生產苯工廠的職工發生白血病的危險是普通人羣的5~6倍,自接觸至發病,即潛伏期,平均爲11.4年。連續吸入高濃度苯的實驗小鼠,80天后11%的雌鼠及19%的雄鼠發生AML。
吸菸者患白血病的危險是普通人羣的2~3倍。菸草中含苯、烏拉坦、亞硝胺,還有放射性物質。吸菸每天超過40支者發生AML後發現有5號或7號染色體異常。
較長期應用烷化劑或鬼臼毒素類的腫瘤和非腫瘤病人發生白血病的危險較正常人羣高出250倍以上。國內銀屑病患者應用乙雙嗎啉、乙亞胺等細胞毒藥物1~7年(平均30個月)後,發生白血病的病例已超過200例,且大多爲AML。
8.2 電離輻射
電離輻射誘發白血病已獲證實。1984年全國26個省、市、自治區調查30年內從事臨牀X線工作者2萬餘人,白血病的標化發生率是對照組的3.5倍,AML佔34.4%。接受X線治療的強直性脊柱炎患者,其白血病發生率爲同年齡組的9.5倍。日本遭原子彈爆炸輻射影響的人羣,白血病發生率爲正常人羣的4~40倍,且和受輻射的劑量成線性關係。上述受輻射人羣發生白血病共766例,其中48%爲AML。多種實體瘤放療後發生白血病的危險增加2倍。
8.3 遺傳
遺傳已被證明是白血病發病的重要危險因素之一,單卵雙胎之一發生白血病後,其同胞在一年內發生白血病的機會是正常人羣的5倍。白血病高危家族中有較高的白血病發生率,是正常家族的16倍。伴特殊染色體異常的遺傳病,如Down綜合徵、Fanconi貧血、Bloom綜合徵、神經纖維瘤病等的白血病發生率遠高於正常人羣。
某些獲得性疾病可轉化爲急性髓細胞白血病,最常見的是骨髓異常增生綜合徵(MDS)轉化爲AML,以往曾將轉化前的MDS稱之白血病前期。由MDS轉化的白血病絕大多數爲AML。其他如真性紅細胞增多症、原發性骨髓纖維化等骨髓增生性疾病在病程後期均有轉化爲AML的可能,少數不典型的再生障礙性貧血、陣發性睡眠性血紅蛋白尿症也可轉化爲AML。
9 發病機制
上述各種可能病因究竟如何促發或轉化爲急性髓細胞白血病,此機制尚不清楚。
9.1 染色體異常
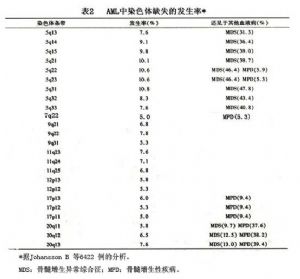
急性髓細胞白血病的染色體異常,像急性淋巴細胞白血病一樣,可分爲2大類:①染色體結構異常,如染色體結構中某一部分缺失(del)、重複(dup)、倒位(inv),或兩個染色體中的某一結構(基因)斷裂,相互易位(t),形成融合基因;②染色體數量的改變,如某一染色體的長臂或短臂缺失(-p,-q),或增加( p, q)。現將AML中已知的染色體異常在AML的發生率,以及見於哪種AML列於表1及表2。

9.2 染色體及基因異常與急性髓細胞白血病分子發病機制的聯繫
大多數急性髓細胞白血病是由於獲得性造血幹細胞或祖細胞的基因突變所致,只有極少數是遺傳或家族性的。造血幹、祖細胞基因突變,多數原因不明。已知的原因有放射線接觸,某些化學物質的作用,尤其是化療藥物如烷化劑,拓撲異構酶Ⅱ抑制劑(如足葉乙甙)等。由於治療所引起的AML稱爲t-AML,近年來報道增多。少數AML的發病機制是由於基因突變加快、DNA修復缺陷、DNA複製錯誤所致。
基因的突變可表現爲染色體的異常,如表1及表2所列的染色體異常,後者的本質是基因組的某一核苷酸序列發生斷裂或突變。
9.2.1 (1)融合基因
在表1中所列舉的染色體異常及其累及的基因中,與AML發病機制研究得較多和了解得比較清楚的基因及其融合基因有以下3種。
①第11號染色體的q23:累及的基因名爲MLL(髓-淋白血病基因)。MLL正常表達於脾、肝、肺、心、腦、T及B淋巴細胞。由於它與果蠅的trithorax蛋白有同源性,故又稱爲HTRX或HRX基因。通過基因相互易位而與MLL融合的基因不下30個。正常時MLL是一種轉錄因子。在AML中,MLL與其配對的基因融合,有的已經克隆。融合基因使MLL的正常基因轉錄調節發生障礙,可能是引起AML及其表型(常見M4、M5型)特點的機制。
②第21號染色體q22:涉及的基因名爲AML1。AML1正常表達在造血細胞。它是核心結合蛋白(CBL)的亞單位,通過一個名爲rhd(runt同源區域)與CBFα形成一種複合物,後者有利於CBF結合在DNA上。AMLl-CBF複合物是一種轉錄因子,與共激活因子ATEF/CREB及P300/CBP以及DNA結合蛋白LEF-1及其接頭的蛋白ALY一起,形成複合轉錄因子,調節IL-3、髓過氧化物酶、T細胞受體、GM-CSF受體(CSF-1R)。這些受體通過AML1結合在DNA上,正常時起轉錄激活作用。若與Groucho或Ear-2蛋白結合,則起轉錄抑制作用。在正常情況下,ETO表達於大腦中的某些細胞、CD34造血祖細胞。在t(8;21)(q22;q22)中,AML1與ETO結合形成融合基因。ETO募集核的共抑制物Sin3A、N-CoR以及與它們結合的組蛋白去乙酰化酶(HDAC),抑制AML1的轉錄激活作用。這一AML1-ETO與核抑制物的複合物,不僅能抑制AML-l的正常功能,而且也抑制ETO的功能,因而擾亂AML-1的轉錄調節作用,這可能是M2b型AML的發病機制。
③維A酸受體α(RARα)及早幼粒細胞白血病(PML)基因。
9.2.2 (2)非融合基因
①p53基因:p53基因定位於人染色體17p13.1,編碼53kD的蛋白。人P53蛋白由393個氨基酸組成,含有4個功能區。野生型P53蛋白是核內的一種磷酸化蛋白,作爲轉錄因子可與特異的DNA序列相結合,一定的外界刺激如DNA損傷、應激等可引起胞內p53蛋白水平升高,激活一系列下游靶基因的轉錄,抑制細胞週期的進行或誘導凋亡。目前已知的靶基因至少有7個。p53基因抑癌功能喪失是惡性腫瘤最常見的現象之一。在血液惡性腫瘤中,p53基因失活與CML急變的關係受到重視。最近有研究者發現CML中p53基因的結構和表達異常,等位基因缺失重組,或點突變,約見於25%的CML急變患者。
②nm23基因:nm23基因存在nm23-H1和nm23-H2兩種亞型,位於人類染色體17q21.3相距4kb。均含有5個外顯子。兩種亞型位於外顯子-內含子連接區的大部分切割位點是一致的。nm23基因編碼一個17kD蛋白。兩種基因亞型編碼的蛋白質分別與核苷二磷酸激酶(nucleosi dediphos phate kinase,NDPK)的A、B亞單位相對應,NDPK影響細胞的發育、增殖、分化及運行調節。而nm23-H1和nm23-H2的一個等位基因失活可能導致NDPK A、B亞單位比例的失衡,引起細胞活動的改變,促進腫瘤的浸潤及轉移過程。nm23基因在一些腫瘤中表達下降與高轉移潛力有關,在血液病中則作爲一種分化抑制因子基因參與疾病的發生、發展過程。但人們尚未確切闡明nm23基因如何參與白血病的發生。促進白血病細胞的增殖和對細胞分化的調控作用。
③BCL-2:BCL-2是控制細胞凋亡基因家族中的一員。定位於人類染色體18q21.3,由3個外顯子組成,編碼229個氨基酸組成的膜蛋白,具有抗凋亡作用, BCL-2可與BAX形成異二聚體,BCL-2/BAX比率是影響細胞凋亡的關鍵,若BCL-2表達高,則抑制細胞凋亡,反之,若BAX表達高,則促進細胞凋亡。體外實驗顯示,BCL-2表達增高能使白血病細胞抵抗糖皮質激素、VP-16、柔紅黴素、米託蒽醌等藥物所誘導的凋亡。同時研究者發現,BCL-2高表達明顯延長白血病細胞生存時間,抑制或阻斷多種因素包括p53、c-myc、化療藥物、撤除生長因子等所觸發的細胞凋亡。另外,BCL-2家族與白血病耐藥有關,高表達BCL-2的白血病細胞對化療藥物不敏感、預後差。
④p16:p16基因是重要的抑癌基因,位於染色體7p21,編碼16kD蛋白,又名多腫瘤抑制基因。p16蛋白抑制細胞週期蛋白依賴性激酶(CDK)4和6,是細胞G1/S期轉換的關鍵調控基因。Hebert等報道p16基因缺失、突變在急性T淋巴細胞白血病(T-ALL)檢出率最高,達22/24,而在前B細胞白血病p16基因缺失檢出率爲11/53。但在AML中批p16基因缺失、結構改變等異常少見,提示在造血系統惡性腫瘤的發生和演變中。p16具有不同的作用。
⑤WT-1:WT-l基因與腎母細胞瘤(Wilm’s tumor,WT)相關。有實驗證實,WT-l是人早期生長反應基因(EGRl)的功能拮抗蛋白,WT-l表達限於腎臟和泌尿生殖系統前體細胞,可能通過阻滯ERGl的促增殖作用,間接促進細胞分化而起抑癌作用。WT-l基因與血液系統惡性腫瘤的關係不甚清楚,但發現白血病細胞常表達WT-l。
⑥其他基因:FMS編碼CSFI受體,其突變及等位基因缺失,可能在某些白血病的發病中具有重要作用,如FMS突變在M5型AML中發生率高。ras基因突變在AML中的發生率可達30%。抑癌基因RB基因失活在各型白血病的發生率爲10%~30%左右。但上述各種單基因異常與AML的發病分子機制之間的關係尚待進一步闡明。
10 急性髓細胞白血病的臨牀表現
兩型急性白血病(ALL和AML)的主要臨牀表現是大同小異,又各有特點。
10.1 貧血
如蒼白,無力,心悸,氣短等,老年病人貧血更爲多見。少數病例可在確診前數月至數年先出現難治性貧血(refractory anemia,RA),以後再逐漸發展成AML(但絕少發展爲ALL)。發生貧血的原因有:由於正常造血幹細胞因白血病克隆增殖而受抑,紅系祖細胞對紅細胞生成素(erythropoietin,EPO)的反應性降低,骨髓微環境破壞,使紅細胞生成減少;出現無效紅細胞生成;合併明顯(少見)或隱性溶血,紅細胞壽命縮短;合併急、慢性失血,或脾功能亢進等。
10.2 發熱和感染
發熱是初診尤其是化療骨髓抑制期患者的常見症狀,其原因主要是感染,感染可發生在體表、體內任何部位。中性粒細胞減少(當<1.0×109/L時感染機會明顯增多)伴功能缺陷,化療和皮質激素的應用使機體免疫功能下降,皮膚、黏膜(口腔、胃腸道等)出血、潰瘍導致屏障破壞是引起感染的主要因素。
10.3 出血
約60%的初診AML有不同程度出血,皮膚黏膜(鼻、口腔及牙齦)出血最常見,眼底、球結膜出血較易見,女性可有月經增多,血尿較少見,但鏡下血尿不易被發現,嚴重的胃腸、呼吸道和顱內出血雖不多見卻常是致死的原因。急性白血病出血的機制較複雜:骨髓衰竭導致血小板減少是最重要的原因。通常血小板<20×109/L時多伴高危出血傾向,若合併全身感染或嚴重貧血時更可加重出血;化療、細菌內毒素和白血病細胞浸潤損傷血管內皮以及凝血障礙都是引起出血的因素。
AML-M3亞型(急性早幼粒細胞白血病)的出血比ALL和AML其他亞型更嚴重而多見,其明顯出血往往與血小板減少的程度不相適應,這是因爲白血病細胞破壞(尤在化療開始後),大量促凝物質和組織因子釋放,可使50%~75%的M3病例發生瀰漫性血管內凝血(disseminated intravascular coagulation,DIC)伴原發纖維蛋白溶解(fibrinolysis),偶爾DIC也出現於AML其他亞型如M5(急性單核細胞白血病)。
10.4 白血病浸潤表現
急性髓細胞白血病髓外浸潤可發生在本病各亞型,但以M5和M4(急性粒單核細胞白血病)爲較頻見。
10.4.1 (1)皮膚浸潤
以M5和M4型多見。外觀呈斑丘疹、結節狀或腫塊,色澤紫紅,可多發而布及全身或少數幾個散佈於體表,且對放療敏感。偶爾也有在血象、骨髓象出現白血病改變前皮膚浸潤先被發現。與AML相關的良性皮膚損害還有多形性紅斑、Sweet綜合徵、膿瘡病、壞疽病等,藉皮膚活檢可資鑑別。
10.4.2 (2)眼部改變
AML視網膜、脈絡膜浸潤比ALL少見,可合併出血或引致失明,眼底浸潤往往提示合併CNS受累。
10.4.3 (3)口腔牙齦改變
25%~50%的M5和M4患者可因白血病浸潤出現牙齦增生,嚴重者牙齦腫脹如海綿狀,表面破潰出血,但AML其他亞型牙齦增生少見。口鼻黏膜、扁桃體或舌體浸潤則較不多見。
10.4.4 (4)肝、脾、淋巴結腫大
肝、脾、淋巴結腫大見於約40%的病例(M5型較多見)與ALL相比其發生率較低,腫大的程度也較輕(肝脾通常肋下剛觸及至肋下<4cm),明顯的肝、脾、淋巴結腫大發生率一般≤10%。對有顯著肝脾腫大的患者應注意與慢性粒細胞白血病(chronic granulocytic leukemia,CGL)急性變相鑑別。
10.4.5 (5)骨關節痛
發生率約20%,比ALL少見。骨關節痛易發生在肋骨、脊椎骨,或肢體長骨及肘、踝等大關節,偶爾可出現骨壞死,但關節滲液稀見。胸骨壓痛是常見體徵,有助於白血病診斷。
10.4.6 (6)中樞神經系統受累(CNSL)
初診AML的發生率不詳,但包括復發時的全病程CNSL總發生率兒童爲5%~20%,成人約15%,明顯低於ALL。年輕(尤其<2歲),周血白細胞和原始細胞數增高,顯著肝脾腫大,M4或M5亞型,以及伴染色體單體7或inv (16)(p13;q22)是發生CNSL的高危因素。生存時間愈長的患者CNS受累的發生率也較高。患者可無症狀,也可表現頭痛等顱內壓增高或腦神經麻痹(V,Ⅶ對腦神經爲主)等症狀。AML的CNS預防性治療一般不作爲常規,但有主張對兒童、M5型或白細胞>100×109/L的患者應給予預防治療。
10.4.7 (7)粒細胞肉瘤
粒細胞肉瘤是由原粒或原單核細胞組成的一種髓外腫瘤,因瘤細胞內含大量髓過氧化物酶使瘤體切面呈綠色,故又名綠色瘤(chloroma),發生率佔AML的2%~14%,且更多見於伴t(8;21)染色體異常的患者。粒細胞肉瘤常累及骨、骨膜、軟組織、淋巴結和皮膚,好發在眼眶、鼻旁竇、胸壁、乳房、涎腺、縱隔、神經、胃腸道和泌尿生殖系等處。瘤塊可於AML診斷時被發現,亦可在AML診斷確立前即出現,並對放療顯示敏感。
AML還可以發生心臟、心包、肺、胸膜、腎及胃腸等各種器官、組織的浸潤,但一般很少導致出現臨牀症狀。可能出現的與浸潤器官相應的症狀、體徵大致有心率不齊,心前區收縮期雜音,心包、胸膜出血和積液,肺X線改變,腎腫大,蛋白尿,尿中出現紅、白細胞,食慾不振,噁心嘔吐,腹痛,腹瀉,胃腸出血或表現爲闌尾炎等。睾丸、前列腺、卵巢、子宮浸潤較少見。
11 急性髓細胞白血病的併發症
11.1 感染
發熱是急性白血病的最常見併發症,約半數以上的患者以發熱起病,當體溫>38.5℃是常常由感染引起的,其熱型不一且熱度不等。發熱的原因是主要細菌和骨骼疼痛。
11.2 出血
急性白血病的整個過程中,幾乎所有的患者都會有不同程度的出血,40%~70%患者起病時就有出血,在未併發DIC時,出血發生率67%~75%,死於出血者佔38%~44%,併發DIC者,幾乎全部有出血,其中20%~25%死於DIC。出血部位以皮膚、黏膜最多見,表現爲皮膚出血點、淤斑、鼻出血、牙齦滲血、口腔舌面血皰和月經過多等,而且淤斑中央常有硬結。嚴重者可有各種內臟出血,如消化道、呼吸道和泌尿道出血,顱內出血常可致命。視網膜出血可致視力減退甚至失明,蛛網膜下隙出血常引起突然死亡,耳內出血可致眩暈,耳鳴、聽力下降等。急性白血病中以AML-M3和AML-M5出血重,易合併DIC。
11.3 白血病髓外併發症
由於白血病細胞可以侵犯各種組織器官,或影響各系統功能,因此可引起多種併發症,有時這些系統併發症甚至成爲患者的主要臨牀表現。可見於成人呼吸窘迫綜合徵、結節病、胸腔積液、肺纖維化,心包積液、心律失常、高血壓、心臟功能衰竭、急腹症、門脈高壓、腎功能不全等。
11.4 血液系統併發症
見於血小板減少、DIC、血栓形成,溶血性貧血、高白細胞狀態與白細胞淤滯綜合徵等。
11.5 內分泌與代謝併發症
11.6 神經系統併發症
顱內出血是白血病患者嚴重併發症,是導致患者死亡的主要原因之一。中樞神經系統白血病AML多見於M4M5型。
11.7 皮膚損害
白血病併發皮膚損害較爲常見,可分爲特異皮膚損害(多與白血病皮膚浸潤有關)和非特異性皮膚損害。特異性皮損表現爲紅斑、結節、腫塊。M5、M3型相對較多。
11.8 骨關節病變
骨關節疼痛是白血病常見的併發症,其他骨髂併發症有顱骨缺損、股骨頭壞死。
11.9 眼部併發症
網膜出血、視盤水腫是白血病患者常見的表現。其他眼部合併症有結膜充血、水腫、前房積膿、脈絡膜浸潤、虹膜浸潤、玻璃體混濁,視力減退、眼眶腫塊、眼球突出、急性青光眼等。主要見於M5型。
11.10 綠色瘤
綠色瘤是AML或CML髓外浸潤的表現,主要由原始或幼稚粒細胞、單核細胞形成有腫瘤,較常見的發生部位爲皮膚、眼眶、其他的部位沿有鼻旁竇、骨、胸壁、乳腺、胃腸道、呼吸道或泌尿道、CNS或淋巴結。T(8;21)AML具有髓外浸潤特點,綠色瘤較多見。一般認爲有綠色瘤的白血病對治療的效果較差,預後不良。
11.11 口腔併發症
11.11.1 (1)牙齦增生
及舌側發展、充血呈海綿狀,質較柔軟。局部可有壞死、出血。化療後牙齦增生可減輕、消失。
11.11.2 (2)口腔黏膜病變
可表現爲出血、糜爛、潰瘍、紅斑、血皰等。與白血病患者血小板減少性出血、口腔感染、化療對黏膜的損傷有關。口腔黏膜病變的重要性在於它可能成爲細菌入侵的門戶。
11.12 白血病相關性副瘤綜合徵
白血病患者併發的某些臨牀綜合徵與白細胞細胞髓外浸潤無關,稱爲白血病相關性副瘤綜合徵(paraneplastic syndromes associated with leukemia)。主要的白血病相關性副瘤綜合徵有Sweets綜合徵、壞疽性膿皮病、關節炎及血管炎綜合徵。
臨牀上伴有皮損及發熱的AML抗生素治療無效,皮損或血培養未發現病原體,應考慮Sweet’s綜合徵。其診斷有賴於皮膚活檢,證明真皮層中性粒細胞浸潤,排除病原體感染、白血病細胞浸潤和血管炎即可確診。
壞疽性膿皮病(pyoderma gangraenosum)是一種病因未明的潰瘍性皮膚病,50%~80%與全身性疾病有關。近1%的壞疽性膿皮病與血液病有關。AML、CML是最常見類型,伴壞疽性膿皮病的ALL和HCL偶見於報道。壞疽性膿皮病可作爲白血病的初診時表現,有些甚至早於白血病的診斷。
12 實驗室檢查
12.1 外周血
外周血白細胞可以正常、升高或減少,各佔1/3的比例。但不論白細胞總數是多少,其中白血病胞佔了85%。有10%~15%的AML病例患病時的外周血白細胞數超過100×109/L,即高白細胞症,多見於M4或M5型的患者,常伴肺部、中樞神經系統浸潤、腫瘤溶解綜合徵和白細胞黏滯症,屬高危型,預後差。極少數患者外周血白血病細胞大於30%,而骨髓中少於30%,未達到急性白血病診斷標準,稱之爲外周血型急性白血病,其中部分病例的骨髓白血病細胞數可能在隨後的幾個月內高,對這些患者尤其老年AML患者,在外周血血小板和粒細胞減少並具有明顯危險性時(血小板<20×109/L,粒細胞<1×109/L),可以暫緩化療。
12.2 骨髓象
多數患者極度增生,正常造血細胞被白血病細胞取代;少數患者骨髓增生低下,但原始細胞仍在30%以上。白血病細胞常有形態異常和核、漿發育不平衡。如胞質內發現Auer小體,更有助於排除ALL而確診爲AML。有時可遇骨髓幹抽現象,原因是白血病細胞極度積聚,致骨髓過分黏稠,或合併骨髓纖維化所致,此時須做骨髓活檢確診。
根據形態學和細胞化學特點,1976年FAB協作組指定了急性白血病分型診斷標準,並於1985年進行了修訂和擴充(表3)。

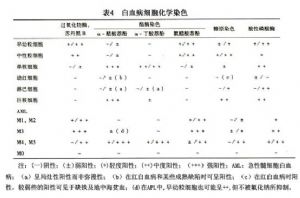
12.3 細胞化學染色
細胞化學染色可補充形態學的不足,在急性白血病的類型鑑別上起着重要作用。常用的細胞化學染色方法包括髓過氧化物酶染化物酶染色(MPO)、蘇丹黑B染色(SBB)、氯醋酸AS-D萘酚酯酶染色(NAS-DCE)、α-丁酸萘酚酯酶染色(α-NBE)、α-醋酸萘酚酯酶染色(α-NAE)、過碘酸-雪夫染色(糖原染色、PAS)、酸陛磷酸酶染色(ACP)、鹼性磷酸酶染色(NAP)、溶菌酶等方法。必要時可做酯酶雙染色和Phl(φ)小體等。根據上述方法,將FAB各型初步分類(表4)。
12.4 電鏡檢查
電鏡檢查通過觀察細胞的超微結構,提高急性白血病形態學分類的正確性。急性粒細胞白血病、急性單核細胞白血病、急性淋巴細胞白血病和巨核細胞白血病的白細胞相互之間的鑑別,可藉助電鏡細胞化學染色來明確診斷。目前電鏡細胞化學染色有MPO染色和血小板過氧化物酶(PPO)等。其優點是靈敏度高,特異性高,能揭示白血病細胞部分早期分化特徵。例如MPO反應對非常幼稚的原粒細胞白血病最具診斷價值,AML的原粒細胞對MPO反應強陽性( ),原粒細胞對MPO的反應不僅限於顆粒,亦見於內質網、核膜和高爾基體;急性單核細胞白血病原始細胞反應弱陽性( ),部分細胞陰性;而急性淋巴細胞白血病和巨核系的原始細胞均無反應。PPO陽性反應是巨核細胞和血小板的特有標誌,巨核細胞對PPO反應爲陽性( ),而急性粒細胞白血病、急性單核細胞白血病和急性淋巴細胞白血病的原始細胞均陰性。
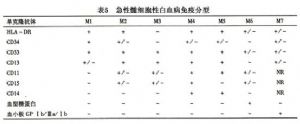
12.5 細胞免疫表型
常用的髓細胞系抗體爲MPO、CD33、CD13、CDllb、CDl5、CD14,其他與髓系相關的抗體是CD34、HLA-DR等,抗血型糖蛋白單抗以及抗血小板糖蛋白Ⅱb/Ⅲa、Ⅰb(CD41a、CD41b、CD61、CD42a、CD42b)分別被認爲是鑑別M6和M7型ANLL敏感而特異的單抗,90%以上M3型ANLL以CD33、HLA-DR爲特點,CDl4是單核細胞特異性抗體,然而敏感性不夠高,在M4和M5型ANLL中,陽性率約佔70%。急性髓細胞系膜標誌與FAB分型關係可見表5。以上髓細胞免疫分型同樣有助於慢性粒細胞白血病急變類型的鑑別。
12.6 細胞遺傳學檢查
通過傳統方法檢測,有50%±10%的AML病例有異常的染色體核型,而染色體顯帶分析,熒光原位雜交(FISH)等細胞遺傳學技術及聚合酶鏈反應(PCR)、Northern、Southern及Western印跡等分子生物學技術的發展和應用,使人們對急性白血病細胞遺傳學和分子生物學有了更深入的認識。染色體改變包括結構和數量的異常(表1,2,6)。
12.6.1 (1)染色體結構異常
①t(8;21)(q22;q22)和inv(16)(p13;q22):是初治AML患者中的最常見的細胞遺傳學異常。t(8;21)佔青年和兒童AML患者發病率的5%~10%,主要與M2型密切相關,同時也可見於M1和M4的病例中。在成人白血病,t(8;21)的存在表明該患者對化療反應良好,緩解率高,中位生存時間長,但在兒童白血病,t(8;21)的存在表明患者對治療反應差。伴隨inv(16)和t(16;16)的AML病例有其獨特的形態學表現:即急性粒單細胞白血病和M4E0,佔成人和青年AML的10%~12%,嗜酸細胞>5%。多數伴inv(16)的AML病例,特別在有AML-M4-E0型樣的形態學改變的預示着有較好的預後,這一異常的染色體表達還可存在於其他的如M2,M4,M5分型的AML病例中。
②t(9;22)(q34;q11):在初治AML的發生率佔1%,t(9;22)通常單獨出現而不伴有複合染色體的改變,但有時伴t(9;22)易位的AML可出現-7及不同的三倍體。
③t(15;17)(q22;q21):是M3(APL)的特異性染色體改變,見於90%以上的M3病例。1%~2%APL患者有t(11;17)易位,個別患者存在t(5;17)易位。具有典型t(15;17)的患者對全反式維A酸(ATRA)有良好的治療反應,而其他二型對ATRA無反應。
④11q23重排:累及11q23條帶重排形式的多見於AML(M4)、ALL、MDS和繼發於曾經接受拓撲異構酶Ⅱ抑制劑而引起的AML。目前發現有大約30種的不同的染色體區帶可與11q23發生易位。例如t(11;19)常見於嬰兒白血病,經誘導分化治療後具有髓系和淋巴系雙表型特徵,說明染色體易位可能發生在多能造血幹細胞階段;在90%以上伴有11q23異常的白血病患者,可以累及不同的基因,一般認爲,累及11q23上的MLL(ALL-1,HRX,Ht-2X)基因通常預後較差。
⑤inv(3)(q2l;q26):伴inv(3)(q2l;q26)的AML是一個獨特亞羣,常伴微小巨核細胞增多和異常血小板增生,這些血液學異常也可見於t(3;3)(q2l;q26)的病例。累及3q異常的血液病患者預後通常較差。

12.6.2 (2)染色體數量異常
①+8:是AML最常見的核型改變,約佔AML患者核型異常的20%。+8作爲原發改變多見於M1、M4和M5,在M3型中較少見,而作爲繼發改變,多見於M3型,也偶見於M2型。
②+21:有1%的發生率,而且多發生於同時有del(5q),-7染色體改變的AML病患。
③-7:在AML患者中有1%~3%的發生率,其出現可能與接觸某些化學物質有關,伴有-7的AML患者對治療不敏感,並代表該疾病的不良預後。
④-5:與毒物接觸有關,常見於治療相關的AML(t-AML)。
⑤性染色體減少:常見於M2型,伴隨t(8;21)。作爲單一核型異常,-X明顯比-Y少。在臨牀上,骨髓細胞-Y往往代表正常男性老年化現象,但有時是腫瘤細胞惟一的核型改變。
12.7 分子生物學檢測
有的融合基因如PML-RARα、AMLl-ETO需要用分子生物學技術纔會予以確診(即所謂MCIM分型),或觀察殘餘白血病。而有的如前文所述,單基因如N-RAS、K-RAs和BCL-2等癌基因和腫瘤抑制基因RB1和p 53等質或量的表達異常可能與某些AML的發生有關。這些基因檢測對預測AML的預後有一定價值。
12.7.1 (1)p53
p53在AML中的表達低,往往預示預後差。
12.7.2 (2)nm-23
nm-23-H1、nm-23-H2mRNA在AML、CML-BC中,水平增高,在CML-CP則正常,nm-23-H1和nm-23-H2在有染色體異常AML-M2和AML-M3中的表達比無染色體畸變的其他類型的AML亞型低;而在AML-M6病例中nm-23表達水平極高。
12.7.3 (3)BCL-2
在AML中,M1、M2亞型的BCL-2表達高於M3、M4、M5,並且高表達者生存時間短,化療效果差。
12.7.4 (4)p16
AML中p16的同源基因p15功能滅活率高達86%,另外,初治和復發AML其骨髓細胞p16表達活性明顯低於正常對照和處於長期緩解AML的骨髓細胞。
12.7.5 (5)WT-1
白血病患者診斷初期的骨髓中的WT-1基因呈高度表達,而當該患者經過治療臨牀達到緩解後,WT-1基因消失,有研究者動態觀察了33例白血病患者(26例AML患者,7例ALL患者)和6例正常人的外周血中的WT-I基因表達,發現6例正常人均未檢測出WT-1轉錄的最低水平(<10-4),而3l例(93.9%)白血病患者的WT-l在疾病初期呈不同水平的表達(10-4~101),而且WT-l基因的表達在AML和ALL無差異,研究者又隨訪檢測了31例獲得CR後的白血病患者的外周血的。WT-1水平表達,共有5例病例復發,(其中2/18例患者並不能檢測出WT-1表達,而3/13患者檢測出低水平的WT-1表達),該5例患者中的3例在復發時WT-1表達再次升高,提示T-1是一敏感的腫瘤標記,能用於監測白血病患者經化療或CR後的微小殘留病竈。
13 輔助檢查
一些AML患者的血清和尿液中的乳酸脫氫酶(LDH)和溶菌酶升高,而這些指標的升高表示着腫瘤相應的負荷量,多見於M5和M4型患者。鈣代謝混亂相對少見,表現爲高鈣血癥或低鈣血癥。可能與白血病細胞產生並釋放一些甲狀旁腺素樣物質有關。
13.1 骨髓病理
白血病患者全身骨髓均有白血病細胞增生浸潤,肉眼可見骨髓呈棕色,或灰白略帶綠色膿樣色澤,如有出血則呈暗紅色或紅褐色,白血病細胞增生嚴重時長骨中的黃髓也可被紅髓所代替(即由白血病細胞增生所佔據)。白血病常伴有骨小梁減少變細或蟲蝕樣缺損,這與骨髓內壓力增高以及骨小梁供血不足有關。網狀纖維增多或膠原纖維增生。
13.2 凝血異常
出現DIC時可出現血小板減少,凝血酶原和部分凝血活酶時間延長,血漿纖維蛋白纖維蛋白原減少,纖維蛋白降解產物增加和凝血因子Ⅴ、Ⅶ、Ⅷ、Ⅹ等的缺乏。
13.3 代謝異常
高尿酸血癥常見於白細胞數增高和誘導化療期患者,且與腫瘤溶解有關,但AML的高尿酸血癥發生率比ALL低;血清乳酸脫氫酶(LDH)可升高,尤其是M4、M5亞型,其增高程度一般也輕於ALL;血清溶菌酶(1ysozyme)增高亦以M4、M5型多見;過量溶菌酶可損傷腎近曲小管,加上白血病本身的代謝異常,抗生素、利尿劑等治療的影響可導致低鉀血癥;高鉀血癥的出現與腫瘤溶解及高尿酸血癥有關;有時還可發生低鈣或高鈣血癥。
14 急性髓細胞白血病的診斷
根據臨牀表現、外周血象、骨髓形態學檢查、急性髓細胞白血病不難診斷。1976年法、英、美三國7位著名血液病專家共同觀察大量急性白血病患者的血片及骨髓片後製訂了FAB分型診斷標準。1985年又進行了修改和補充,以後又有小的修正,目前此標準已爲世界各國所接受。我國在1986年也按FAB分型診斷標準修訂了國內標準,基本上和國際標準一致。下面介紹AML的FAB分型診斷標準要點,及各型其他特點。
14.1 急性粒細胞白血病未分化型(M1)
骨髓中原始細胞Ⅰ型(典型原粒細胞、胞質中無顆粒)+Ⅱ型(有原粒細胞特徵,胞質量少,有少量細小顆粒)>90%,早幼粒細胞少,中性中幼粒細胞及以下階段粒細胞不見或罕見,至少3%的原始細胞過氧化物酶或蘇丹黑染色陽性。M1型佔AML的10%~20%,年齡中位數40~50歲,僅1/3有肝、脾或淋結腫大。血象大多呈紅細胞及血小板減少,半數白細胞增多,1/4白細胞減少。無特殊的細胞遺傳學異常,通常對化療敏感,預後較好。
14.2 急性粒細胞白血病分化型(M2)
骨髓原始細胞I+Ⅱ型佔30%~89%,早幼粒細胞及以下階段粒細胞>10%。M2型佔AML的30%~45%,平均年齡爲30歲。常見細胞遺傳學異常,其中29%~40%爲t(8;21),且Auer小體常陽性。免疫表型除具髓系特點外,可伴CD56及CD19陽性。
t(8;21)累及二個基因,即AML1(21q22)及ETO(8q22),二者形成融合基因AMLl/ETO,在長期CR者仍可檢出,故不宜作爲復發的指標。t(8;21)主要發生於無MDS病史的M2型,治療反應好,CR率高,長生存期者多,但兒童患者、伴髓外病變者仍有較高的複發率,壽命較短。男性M2患者常伴Y染色體丟失,女性常伴X染色體丟失。
14.3 顆粒增多的早幼粒細胞白血病(M3)
顆粒增多的早幼粒細胞白血病(M3)又稱急性早幼粒細胞白血病(APL)。骨髓早幼粒細胞>30%。如胞質顆粒粗大、密集或融合,稱粗顆粒型(M3a);如顆粒細小而密集,稱細顆粒型(M3b);如周圍血早幼粒細胞顆粒甚少或缺如,而骨髓中仍爲典型的早幼粒細胞,稱變異型(M3v)。各型Auer小體均多見。
APL佔AML的5%~10%,病人常較年輕,年齡中位數30~38歲,10歲以下者罕見,歐洲、中南美洲的拉丁裔民族發病較高。90%的病人表現有繼發DIC的出血,系白血病細胞顆粒釋放促凝物引起。部分病人釋放促纖溶物質,致纖溶亢進而出血。但自從應用全反式維A酸(ATRA)後,出血、特別是嚴重出血者已少見。外周血白細胞常常減少,且大多爲M3a,而白細胞升高者多見於M3b及M3v。早幼粒細胞由於有大量顆粒,有時還伴大量柴束樣的Auer小體,使細胞核觀察不清,故又稱爲“霧細胞”。
染色體17q21含有維A酸受體(RAR)α基因,而15q24是早幼粒細胞白血病基因(PML)所在的位置,95%以上的早幼粒細胞白血病發生t(15;17),所形成的融合基因有兩種形式:①PML/RARa,位於15P及它的互補位置。②RARα/PML,位於17P-。前者見於所有的M3型患者,後者則見於2/3的M3型患者。PML 在 15號染色體基因斷裂點有3種,分別爲長型、短型及變異型。長、短型皆對ATRA治療反應好,但短型的預後仍差於長型,而變異型對ATRA敏感性差,且常伴其他的細胞遺傳學異常,預後最差。APL還有非t(1 5;17)的其他細胞遺傳學異常,如t(5;17)(NPM/RARα、t(11;17)(PLZF/RARa)。此兩種類型的APL對ATRA耐藥,預後差。
14.4 急性粒-單核細胞白血病(AMMOL,M4)
粒-單系二種細胞以不同比例同時存在於骨髓和周圍血中,包括:①M4a:原始和早幼粒細胞增生爲主,原、幼單核和單核細胞>20%;②M4b:原、幼單核細胞增生爲主,原始和早幼粒細胞>20%;③M4c:原始細胞既呈粒細胞系、又呈單核細胞系形態特徵者>30%;④M4E:除上述任一項條件者,同時存在5%~30%的細胞伴粗大而圓的嗜酸顆粒及着色較深的嗜鹼顆粒,前者電鏡下無中心晶體樣結構。
AMMOL佔AML的5%~10%,年齡中位數40~45歲,肝脾及淋巴結腫大多見。血白細胞大多升高。其中20%~25%>100×109/L。CNS-L、齒齦及皮膚浸潤多見。
M4E幾乎均有inv(16)(p13;q22),導致編碼平滑肌肌球蛋白鏈基因(MYH11)和編碼核結合因子β單位基因(CBFβ)發生融合,即MYH11/CBFβ10%的無嗜酸細胞增多的M4也可檢出MYH11/CBFβ長期臨牀CR的M4E患者,其MYH11/CBFβ仍可存在。
M4E易累及CNS,故必須採取預防措施,全身用大劑量阿糖胞苷可降低CNS-L的發生率。M4ECR率高,預後相對較好。
少數M4型合併血嗜鹼粒細胞增多,骨髓常有三系病態造血及環狀鐵幼粒細胞,伴t(6;9),預後差。
14.5 急性單核細胞白血病(AMOL,M5)
骨髓原始單核細胞≥80%,稱未分化型,即M5a。原始單核細胞<30%,稱分化型,即 M5b。
AMOL佔AML的2%~10%,M5a患者年齡偏小,75%<25歲。M5無特異性的染色體異常,但常累及第11號染色體,如t(11;9)、t(11;17)、t(11;19)、11q23平衡易位,與混合白血病(MLL)基因有關,MLL基因的氨基端分別和9號染色體的AF9基因及19號染色體的DNL基因融合。
50%的AMOL有髓外病變,包括CNS、皮膚及齒齦等,肝脾腫大多見。CNS-L發生率爲3%~22%。血白細胞常明顯升高,10%~30%伴白細胞血癥。DIC的發生率也較高,以往僅次於APL,ATRA應用後DIC的發生率可能已躍居AML的首位。部分AMOL患者可伴蛋白尿,甚至腎功能不全,可能與血清溶菌酶水平升高而損傷腎有關。AMOL的CR期較短,預後差。
14.6 紅白血病(M6)
當骨髓中紅系細胞>50%,或紅系細胞>30%,但其中15%以上爲形態異常的幼紅細胞,上述兩種情況之一伴原粒細胞或原始單核細胞≥30%(非紅系細胞計數即可),即爲紅白血病。幼紅細胞常伴胞質空泡、核異常及類巨幼變。
M6佔AML的5%以下,年齡多>50歲,男性多於女性,幾乎均有明顯的貧血及血小板減少。不少病例屬繼發性白血病,包括從MDS轉化而來,故預後差。有報告1/3病例有骨痛,部分骨痛病例抗核抗體、類風溼因子及抗人球蛋白試驗陽性,可伴高丙種球蛋白血癥。
14.7 急性巨核細胞白血病(AMKL,M7)
骨髓原始巨核細胞≥30%時,並經免疫分型或電鏡血小板過氧化物酶染化物酶染色陽性證實。若骨髓“幹抽”,有骨髓纖維化,則需行骨髓活檢,經免疫組化證實有原始巨核細胞增多。
M7佔AML的5%以下,是AML中最少見的類型,臨牀表現和其他AML相似。肝脾及淋巴結腫大少見。周圍血細胞常減少,但30%患者的血小板>100×109/L,血小板聚集功能降低。血清乳酸脫氫酶(LDH)常明顯升高。部分病例放射學顯示骨硬化及骨溶解,此在急性白病中罕見。
14.8 M0亞型
此外,尚有一M0亞型,血及骨髓中出現原始細胞,無Auer小體,過氧化物酶染化物酶染色陰性,難以診斷爲AML,但免疫表型檢查有髓系表型,CD13、CD33陽性,髓過氧化物酶陽性,CD34也陽性,表明白血病細胞來自髓系。細胞遺傳學檢查常伴5q-、或7q-。患者白血病細胞常有多藥耐藥基因表達,化療反應較差。
15 鑑別診斷
需和AML鑑別的有下列疾病:
15.1 急性淋巴細胞白血病(ALL)
臨牀上二者相似,僅症狀和體徵的頻度和程度上有所差異,如浸潤表現ALL更爲常見及顯著。形態學檢查可區分大部分AML和ALL,困難者加做細胞化學檢測,絕大多數病例可確診。少數病例需行免疫表型檢測鑑別,僅極少數病例還需進一步經細胞遺傳學(或)和分子生物學檢測。
15.2 類白血病反應
常見的類白血病反應表現爲血白細胞升高,伴少數中、晚幼粒細胞,骨髓顯示粒系左移,因此類似慢性粒細胞白血病。少數類白血病反應,血液學特點爲全血細胞減少,血片中出現原始細胞,骨髓原始細胞也明顯增多,甚至>30%,稱之類急性白血病反應,鑑別點爲有原發病(各種嚴重感染,粒細胞缺乏症恢復期等)、血中性粒細胞鹼性磷酸酶染色積分明顯升高、原始細胞短期內數量有明顯波動,且無Auer小體、血液學改變隨原發病好轉、控制而逐漸恢復正常。
15.3 再生障礙性貧血(AA)
主要和非白血病性白血病及低增生性AML鑑別。根據AML浸潤的臨牀表現及骨髓檢查(包括活檢)不難區分。
15.4 傳染性單核細胞增多症(IM)
IM有和急性白血病類似的臨牀表現,如發熱、肝脾及淋巴結腫大,血片中如有較多的異常淋巴細胞,有時和ALL或AML可混淆。通常經檢查血清EB病毒標誌物、嗜異性凝集試驗及骨髓象可鑑別。此外,IM病程有自限性,4周左右即恢復正常。
15.5 惡性組織細胞病(MH)
此外,AML有時尚需和全血細胞減少的巨幼細胞貧血鑑別,尤其是M6型,因爲二者骨髓中紅細胞系均有巨型變。根據AML骨髓中>30%的原始細胞存在,及葉酸、VitBl2治療3~4周無效,可明確區分。
16 急性髓細胞白血病的治療
急性髓細胞白血病各亞型中,除APL之外,治療基本相同。
16.1 誘導緩解化療
急性髓細胞白血病的經典誘導化療是DA方案:柔紅黴素(DNR )45~60mg/(m2·d)(第1~3天)+阿糖胞苷(Ara-C) 100mg/(m2· d) (第1~5天或第1~7天),第一療程完全緩解率(CR)爲40%~50%;第二療程達60%~75%。
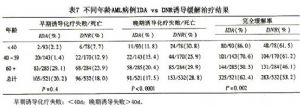
其他誘導化療方案如下:①AID 方案:阿糖胞苷(Ara-C)+伊達比星(去甲氧柔紅黴柔紅黴素)或阿糖胞苷(Ara-C)+伊達比星(IDA)+依託泊苷(VP-16)(ICE);②阿糖胞苷(Ara-C)+柔紅黴素(DNR)+硫鳥嘌呤(6-TG);③米託蒽醌(NVT)+依託泊苷(VP-16)(ME)等。臨牀研究表明,在DA方案基礎上加用依託泊苷(VP-16)並不進一步提高完全緩解率,但可以提高總生存率。而加用硫鳥嘌呤(6-TG)對完全緩解率和總生存率無顯著意義。伊達比星(IDA)在誘導化療中的作用逐漸得到認可,其優勢在於惡性腫瘤細胞毒性強,尤其對伴多藥耐藥表型白血病細胞作用強於柔紅黴素(DNR),心臟毒性低。單療程緩解率高,並且適用於老年患者。標準用法如下:12mg/m2,第1~3天。近來ACG組(AML collaborative Group)總結了5項大規模的臨牀隨機對照研究,共1052例AML。結果伊達比星(IDA)與柔紅黴素(DNR)組比較早期治療失敗/死亡率接近,而晚期治療失敗/死亡率則明顯減低(P<0.0001);總體緩解率伊達比星(IDA)組高於柔紅黴素(DNR)組(P=0.002)。伊達比星(IDA)組無病生存(DFS)和總生存率均略高於柔紅黴素(DNR)組(P=0.07和P=0.03,表7)。因此,伊達比星(IDA)已經逐步成爲臨牀研究中標準的誘導化療方案之一。
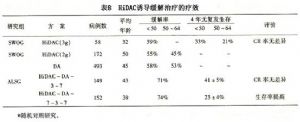
阿糖胞苷(Ara-C)是誘導化療方案中重要的組成部分,其常規劑量爲100mg/m2第l~7天,臨牀研究表明,7天療程效果優於5天療程,而與10天療程相近;持續靜脈點滴優於單次、分次注射;200mg/m2並不提高療效。近年來,由於中-大劑量阿糖胞苷(Ara-C)在AML緩解後治療取得明顯效果,因此部分學者嘗試應用大劑量(HiDAC)進行誘導緩解治療。部分研究提示在年齡<50歲的AML病例可取得近90%的CR率,而且與常規劑量比較,能進一步延長患者的DFS(表8)。因此目前美國腫瘤協作網(NCCN)推薦HiDAC作爲AML誘導緩解的方案之一。
16.2 緩解後治療
誘導治療達到CR後,大劑量鞏固和強化治療在AML的後續治療中有重要的地位,它在很大程度上將決定AML的持續緩解時間,患者生存率及復發的時間。目前主張緩解後治療應該是強烈的鞏固治療,這些方案的強度至少與誘導緩解治療方案相同。應用這樣的方法,中位CR期達18~24個月,20%~45%達CR的患者無病生存期達15年,主要方法如下。
16.2.1 (1)定期強化治療3年方案
阿糖胞苷(Ara-C),100mg/m2十硫鳥嘌呤(6-TG)100mg/m2,12 h重複一次至骨髓抑制,每個月重複。一個治療療程約10天,12~18個月後7天。適用於各年齡段患者,長期DFS預期>10%~20%。
16.2.2 (2)短程大劑量鞏固化療方案
阿糖胞苷(Ara-C),2~3g/m2靜脈注射3h;12h重複一次;第1、3、5天;每28~35天重複或根據外周血計數恢復程度調整。5年DFS預期44%;治療相關死亡5%;適用<45歲AML;45~60歲神經毒性大(12%);>60歲療效差且副作用大。目前以大劑量阿糖胞苷(Ara-C)爲基礎,加或不加用其他藥物(常規劑量),如柔紅黴素(DNR)、伊達比星(去甲氧柔紅黴柔紅黴素)、依託泊苷(VP-16)、米託恩醌等。
16.3 造血幹細胞移植
16.3.1 (1)異體骨髓和造血幹細胞移植(Allo-BMT或Allo-HSCT)
Allo-BMT或Allo-HSCT是目前惟一能根治白血病的方法。臨牀實踐已經表明,應用Allo-BMT/Allo-HSCT治療AML可有效控制疾病的復發,長期DFS在40%~55%。與療效關係密切的因素是病例和供者的選擇和骨髓移植的時機。國外多組單中心研究表明,同胞供者HLA相合的移植,第一次完全緩解期進行,長期DFS(以下簡稱DFS)達45%~70%;復發早期(ER,early relapse)或第二次完全緩解期進行,DFS 20%~35%;難治/復發病例DFS 10%~15%;誘導緩解治療無效病例行移植,DFS 21%~43%。關於移植治療和化療的前瞻性研究提示,Allo-HSCT治療的優勢在於減少復發,提高DFS(表9)。
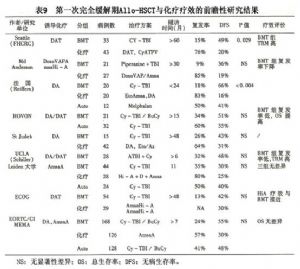
鑑於AML(APL除外)迄今複發率甚高,即使已規劃3~5年DFS仍只有20%左右,因此對這些患者,在獲得CR1後,只要年齡及其他條件許可,原則上應儘可能爭取進行HSCT治療。
16.3.2 (2)自體骨髓/造血幹細胞移植(Auto-BMT/Auto-HSCT)
Auto-HSCT適用於多數AML病例(<60歲),且移植相關併發症和死亡率低,長期生存可達到35%~50%。但Auto-HSCT缺乏GVL效應,主要的缺陷是複發率高。Zittourn總結了1986~1993年由歐洲59個研究中心參加的隨機對照研究,共941例AML病人。平均隨訪時間40個月。其中完全緩解623例(CR率66%),576例接受了一療程的強化治療。168例接受Allo-BMT治療,254例患者進行隨機分組:128例接受ABMT,126例接受第二療程強化療。可評價病例中Allo-BMT l44例,ABMT 95例,化療104例。結果發現,3組病例生存期無顯著差異:Allo-BMT組4年總生存(OS)59%,化療組46%,ABMT組56%(P=0.43)。複發率比較化療組(57.1%)高於ABMT組(40.6%)和Allo-BMT組(24.4%),而治療相關死亡率則Allo-BMT(17.3%)高於ABMT(9.4%)和化療組(7.1%)。無病生存率化療組30%,ABMT組48%,Allo-BMT組55%(P=O.04)。自體骨髓移植淨化一直是自體骨髓移植治療的研究重點之一。Linker CA等報告了應用四氫過氧環磷酰環磷酰胺(4-HC,100mg/ml)處理自體骨髓細胞治療的長期隨訪結果。第一次完全緩解期AML共50例,預處理方案爲白消安(busulfan)16mg/kg+依託泊苷(足葉乙甙)60mg/kg。隨訪時間平均6.8年(最少4.5年)。結果治療相關死亡2例,復發13例,DFS 70%,複發率27%,總生存率爲72%。這是目前隨訪時間最長和療效最佳的一組報告,而其他的一些研究卻未能表明體外淨化處理移植的優越性。主要因素在於不同的研究組在病例選擇,淨化藥物等多方面有較大差異,而淨化體系效率缺乏標準化的檢測手段,因此目前淨化效應仍是有待進一步研究的課題。
16.4 特殊類型的治療
M2b誘導分化治療:AML-M2b患者90%伴t(8;21)特異性染色體改變,形成AML-ETO融合基因。近年來,國內外學者進行了大量的實驗研究,以探討M2b的誘導分化和凋亡療法,研製新的誘導分化劑。苯丁酸鈉對許多腫瘤細胞如HL-60(人早幼粒細胞白血病)、MEL(小鼠紅白血病)有抑制增殖、促進分化作用。美國國立健康研究院的一組研究者發現維A酸(ATRA)與苯丁酸鈉使用有協同作用,故認爲苯丁酸鈉可能有應用前途。目前M2b的誘導分化療法尚處於實驗階段,臨牀療效尚待肯定。
16.5 難治及耐藥AML的治療
按目前急性髓細胞白血病治療水平,仍有10%~30%的患者對一線標準誘導方案無效,40%~80%已經獲得CR的患者最終還要復發。
難治(refractory)和復發(relapsed)急性髓細胞白血病對再治療的耐藥程度和治療反應是各不相同的,並取決於疾病本身的異質性、復發的性質、時機和次數,尤其是初次緩解(CR1)期的長短。凡一線方案充分治療無效,CR後6個月內復發,或復發後經再治療不能達到二次緩解(CR2)的患者屬於高度耐藥的AML。文獻報道CR1<1年和≥2年的初次復發者,使用原標準方案再誘導的CR2率分別爲30%~50%和50%~60%,其中CR1≥2年者,3年DFS可達20%~25%;反之,CR1<1年者的CR2率僅10%~30%,3年DFS爲0%。爲區分高度耐藥的難治病例與一般復發病例,使不同作者報道的治療結果具有可比性,各國學者討論了難治性AML的各種判斷標準,其中德國AMLEG協作組提出的四項標準最爲通用,即:①標準方案誘導化療2療程不緩解;②CR1後6個月內復發;③CR1 6個月後復發,且原誘導方案再治療無效;④二次和多次復發。需要說明,因誘導化療劑量不足導致治療無效者,可能對標準劑量方案依然敏感,並能獲得緩解,這類病例並不屬於難治性AML。
爲克服臨牀耐藥,難治和復發AML的治療選擇是:①使用與一線治療無交叉耐藥的其他藥物組成的新方案;②使用HD、ID阿糖胞苷(AraC);③應用耐藥逆轉劑;④採取造血幹細胞移植。
HD、ID阿糖胞苷(Ara-C)爲主的各種聯合方案是難治和復發AML最常用的挽救治療方案。通常與HD、ID阿糖胞苷(Ara-C)聯合使用的藥物有米託蒽醌(MTZ)、伊達比星(ida)、依託泊苷(VP-l6)、安吖啶(m-AMSA)和門冬酰胺酶等,報道CR率30%~70%,但中位DFS一般≤6個月,3年生存率僅7%。米託蒽醌(MTZ)+HD、ID阿糖胞苷(Ara-C) 依託泊苷(VP16)是近年探索較多療效相對較好的難治、復發AML治療方案,報道CR率大都在50%以上,對CR1<6個月的早期復發者也有較高療效。Amadori、Paciucci和Spadea等認爲阿糖胞苷(Ara-C)與米託蒽醌(MTZ)有時間依賴性協同作用[用藥順序應是先給VP-16,繼之爲阿糖胞苷(Ara-C),最後用米託蒽醌(MTZ)]。伊達比星(Ida)+HD阿糖胞苷(AraC)獲得的CR2率可能較高,但CR2期似並不更長。氟達拉濱是腺苷類藥物,與阿糖胞苷(Ara-C)有協同作用,可提高細胞內阿糖胞苷(Ara-C)活性成分Ara-CTP的濃度。而G-CSF可增加靜止期細胞對細胞毒藥物的敏感性。一些作者採用FLAG方案[氟達拉濱25~30mg/m2×5天,阿糖胞苷(Ara-C)2g/m2×5天,G-CSF 5μg/kg與化療同時起用直至中性粒細胞恢復],使難治、復發AML的CR率達50%~75%,CR和生存期分別爲9.9和13個月,是近年報道療效較好的方案之一。FLAG再加蒽環類能否進而改善療效也在觀察中。鑑於HD阿糖胞苷(AraC)(3g/m2×12天)有嚴重CNS、肝臟、骨髓抑制等毒性,老年患者耐受更差,有主張改用ID阿糖胞苷(AraC)[(0.5~2)g/m2×(6~8)次],認爲可降低治療相關死亡率,而總療效(CR率和生存率)沒有差別。
難治和復發AML的非HD阿糖胞苷(AraC)治療方案有兩類:一類是標準劑量阿糖胞苷(AraC)米託蒽醌(MTZ)±依託泊苷(VP-16)、安吖啶(m-AMSA)或伊達比星(Ida)等;另一類不含阿糖胞苷(AraC),如依託泊苷(VP-16)聯合米託蒽醌(MTZ)、安吖啶(m-AMSA)、阿柔比星(阿克拉黴素)或阿扎胞苷(5-azacytidine)等。兩者CR率大都≤50%,緩解期更短。Brown等採用HD依託泊苷(VP-16)(總量1.8~4.2g/m2)加環磷酰胺HD(CTX)[50mg/kg×(3~4)天],難治AML獲CR率42%,其中曾用HD阿糖胞苷(AraC)治療證明耐藥的病例,CR率也達30%,但主要毒性有黏膜炎,肝損害和出血性膀胱炎、且17%的患者死於骨髓抑制期合併嚴重感染。其他治療還有卡鉑、2-氯脫氧腺腺腺苷(2-chlorodeoxyadenosine,2-CdA)等。
影響復發患者療效的因素除CR1期長短外,還有一線誘導和緩解後治療強度。通常接受過強烈初次誘導及緩解後治療的復發者對再治療的反應將明顯下降;緩解後於治療中復發比完成並停止治療後復發療效更差;二次及多次復發不僅再緩解少見,緩解期也一次比一次縮短,最終難免死亡。影響復發患者療效的其他不良因素尚有高齡(>50歲)、高白細胞數(>25×109/L)、白血病發病前有MDS等前驅血液病史,血清膽紅素和鹼性磷酸酶升高,以及某些高危細胞遺傳學異常等。
由於難治和復發AML單用化療的遠期效果都很差,一般主張對年齡<55歲,有合適供者的原發難治患者和CR1<1~2年的復發病例採用異基因骨髓移植(Allo-BMT)。國際骨髓移植登記處(IBMTR)比較復發AML在CR2後使用Allo-BMT和繼續單純化療的3年LFS,年齡<30歲,CR1≥1年的患者分別爲41%和17%,年齡>30歲,CR1<1年的患者分別爲18%和7%,顯然Allo-BMT的療效要明顯優於單用化療。
16.6 其他正在探索的新方法
實驗證明拓撲異構酶工抑制劑拓撲替康可特異性與DNA單鏈斷端上的拓撲異構酶Ⅰ結合,阻止拓撲異構酶Ⅰ對單鏈斷端的修復,致DNA雙鏈結構破壞,導致細胞死亡,因而具有抗腫瘤活性。Ⅰ期臨牀試驗顯示部分難治、復發AML單用拓撲替康可獲CR,主要毒性是骨髓抑制和黏膜炎。拓撲替康(1~7mg/m2連續5天靜脈輸注)與含阿糖胞苷(AraC)標準方案及拓撲異構酶Ⅱ抑制劑聯合,可能有增強抗白血病的作用,拓撲替康與環磷酰胺(CTX)、阿糖胞苷(AraC)、依託泊苷(VP-16)的聯合治療也在探索中。
MDR1基因過度擴增致細胞膜上P糖蛋白(P-gp)高表達是白血病細胞多藥耐藥(MDR)的主要機制,也是導致AML化療失敗或早期復發的重要因素。某些非細胞毒藥物如環孢素A、環孢素D類似物PSC833有抑制MDRl/P-gp表達,延緩肝臟對葸環類和依託泊苷(VP-l6)的代謝、清除,維持體內化療藥有效濃度的作用,因此聯合使用環孢素(CsA)和化療有助改善這類病例的療效。List等報道環孢素(CsA)聯合柔紅黴素(DNR)+HD阿糖胞苷(ARAC)治療原發難治和CR1<1年的復發AML,CR率67%。PSC833無免疫抑制作用及腎毒性,而對MDR1/P-gP的抑制效應比環孢素(CsA)強10倍,目前其療效正在臨牀評價中,但有關環孢素(CsA)和PSC833的確切療效機制仍待進一步闡明。
單克隆抗體(MoAb)治療白血病近年獲相當進展。由於>90%的AML表達CD33,而正常造血幹細胞不表達,因此CD33是AML治療較理想的靶抗原。抗CD33和抗CD45 MoAb還適於攜帶毒素、藥物或放射性核素,以更有效地清除體內白血病細胞。目前研究較多的有HuMl95(未結合的抗CD33 MoAb)、CMA676(抗CD33 MoAb與抗腫瘤抗生素Calicheamicin的交聯物)和131碘(131I),90釔(90Y),213鉍(213Bi)標記的抗CD33、抗CD45 MoAb(如131I-HuMl95、213Bi-HuMl95、131I-抗CD45)等,臨牀使用這些MoAb治療難治和復發AML,部分病例可獲CR,或見骨髓幼稚細胞減少,而全身毒性較輕。鑑於有效病例大都治療前骨髓白血病細胞<30%,因此認爲MoAb可能對白血病細胞低負荷患者較有效,故還可用於AML鞏固治療後清除體內MRD。MoAb治療AML-M3型的效果更滿意,在ATRA誘導緩解後,應用化療和HuM195(3mg/m2,每週2次),可使本病PML/RARα基因早期轉陰,且轉陰率高,患者的DFS顯見延長。
採用去除T細胞的造血幹細胞移植患者複發率高,間接證明供者T細胞介導移植物抗白血病(GVL)效應。1990年Kolb等首先發現供者淋巴細胞輸注(DLI)有抗腫瘤作用。以後Collins等以DLI治療Allo-BMT後復發的各種白血病,報道CML慢性期患者CR率73%,其中細胞遺傳學/分子生物學復發者CR率100%,但DLI對Allo-BMT後復發的AML療效較差,CR率僅15%~29%,而有效患者隨後常出現髓外復發,這可能與AML的增殖活性,內源性耐藥和白血病負荷高有關。DLI治療的副作用主要是發生GVHD和骨髓抑制,是導致感染、死亡的常見原因。有人從小劑量開始,採用劑量遞增,分多次給予DLI(D3 T細胞從6×l06/kg漸增至1×108/kg),或採用體外去除細胞毒CD8 T細胞,分離CD4 T細胞進行DLI,結果可明顯減少GVHD的發生率,減輕GVHD的嚴重程度。
16.7 療效標準
16.7.1 (1)完全緩解(CR)
①臨牀無白血病細胞浸潤所致的症狀和體徵,生活正常或接近正常。
②血象:Hb≥100g/L或≥90g/L(女及兒童),中性粒細胞絕對值≥1.5×109/L,血小板≥100×109/L。外周血白細胞分類中無白血病細胞。
③骨髓象:原粒細胞Ⅰ+Ⅱ型(原始單粒+幼稚單核細胞或原始淋巴+幼稚淋巴細胞)≤5%,紅細胞及巨核細胞系正常。
M2b型原粒細胞+早幼粒細胞≤5%,中性中幼粒細胞比例正常範圍。
M5型原單核Ⅰ型+Ⅱ型及幼稚單核細胞≤5%。
M7型粒細胞、紅細胞二系正常,原巨核細胞+幼稚巨核細胞基本消失。
16.7.2 (2)部分緩解(PR)
骨髓原粒細胞Ⅰ型+Ⅱ型(原單核+幼稚單核細胞或淋巴母細胞+幼稚淋巴細胞)>5%而≤20%;或臨牀、血象項中有一項未達完全緩解標準者。
16.7.3 (3)復發
白血病復發有下列三者之一者稱爲復發
①骨髓原粒細胞Ⅰ型+Ⅱ型(原單 幼單或原淋巴+幼淋)>5%又≤20%,經過有效抗白血病治療一個療程仍未能達到骨髓象完全緩解者。
②骨髓原粒細胞Ⅰ型+Ⅱ型(原單+幼單或原淋+幼淋)>20%者。
16.7.4 (4)持續完全緩解(CCR)
指從治療後完全緩解之日起計算,其間無白血病復發達3~5年以上者。
16.7.5 (5)長期存活
急性白血病自確診之日起,存活時間(包括無病或帶病生存)達5年或5年以上者。
16.7.6 (6)臨牀治癒
指停止化學治療5年或DFS達10年者。
說明:凡統計生存率,應包括誘導治療不足一療程者;誘導治療滿一個療程及其以上的病例應歸入療效統計範圍。
17 預後
某單一因素常不能可靠地判斷預後,應分析患者的全部信息,才能作出較爲準確的推測。重要的預後因素如下:
17.1 年齡
老年(>60歲)及2歲以下的嬰幼兒預後差。經充分治療,15~60歲者5年無病生存率爲10%,2~14歲的兒童則爲60%。
17.2 繼發性急性髓細胞白血病
如由MDS轉化而來,或因其他良、惡性疾病經化、放療後的AML,化療反應差,或雖獲CR,但CR期短。
17.3 細胞遺傳學
在判斷預後中有重要價值,t(15;17)的APL對ATRA反應好,致DIC已大爲減少,CR後繼續強聯合化療,約50%的病人可長期存活。有t(8;21)的M2型,CR率高,但如合併髓外病變,預後則差。inv(16)的M4E0型,CR率也較高,但易併發CNS-L,影響其預後,近經充分的HD-Ara-C治療,預後已有改善。
繼發性白血病常伴5.7號染色體異常,預後不良。3倍體8是AML染色體數量異常的最常見類型,預後差。伴複雜染色體異常的AML預後極差。
17.4 FAB分型
M0、M5、M6、M7型預後較差;原始細胞伴Auev小體、骨髓嗜酸細胞增多者預後較好。
17.5 治療後骨髓反應
標準化療方案後1周,至多2周達骨髓增生低下;一療程即獲CR者預後好。
17.6 免疫表型
AML的免疫表型對預後的影響報告不一,CD34和p170同時陽性者易耐藥而預後不良。AML伴淋巴系免疫表型,尤其僅伴某一系淋巴細胞表型者預後不良。
17.7 伴高白細胞血癥及髓外病變者預後較差
急性髓細胞白血病的死亡原因,依次爲感染(70%)、出血(15%)、CNS-L(5%)、肝或腎功能衰竭(5%),以及貧血、全身衰弱。