3 註解
藥物代謝動力學,簡稱爲藥動學,研究藥物體內過程及體內藥物濃度隨時間變化的規律。藥物在體內雖然不一定集中分佈於靶器官,但在分佈達到平衡後藥理效應強弱與藥物血漿濃度成比例。醫生可以利用藥動學規律科學地計算藥物劑量以達到所需的血藥濃度並掌握藥效的強弱久暫。這樣可以比單憑經驗處方取得較好的臨牀療效。
4 藥物體內過程
4.1 吸收
藥物的吸收(absorption)是指藥物自體外或給藥部位經過細胞組成的屏蔽膜進入血液循環的過程。多數藥物按簡單擴散(simple diffusion)物理機制進入體內。擴散速度除取決於膜的性質,面積及膜兩側的濃度梯度外,還與藥物的性質有關。分子量小的(200D以下),脂溶性大的(油水分布係數大的),極性小的(不易離子化的)藥物較易通過。藥物多是弱酸性或弱鹼性有機化合物,其離子化程度受其pKa(酸性藥物解離常數的負對數值)及其所在溶液的pH而定,這是影響藥物跨膜被動轉運,吸收分佈排泄的一個可變因素。按Handerson-Hasselbalch公式:

由此可見不論弱酸性或弱鹼性藥物的pKa都是該藥在溶液中50%離子化時的pH值,各藥有其固定的pKa值。當Pka與pH的差值以數學值增減時,藥物的離子型與非離子型濃度比值以指數值相應變化。非離子型藥物可以自由穿透,而離子型藥物就被限制在膜的一側,這種現象稱爲離子障(ion trapping)。例如弱酸性藥物在胃液中非離子型多,在胃中即可被吸收。弱鹼性藥物在酸性胃液中離子型多,主要在小腸吸收。鹼性較強的藥物如胍乙啶(pKa=11.4)及酸性較強的藥物如色甘酸鈉(pKa=2.0)在胃腸道基本都已離子化,由於離子障原因,吸收均較難。pKa小於4的弱鹼性藥物如安定(pKa=3.3)及pKa大於7.5的弱酸性藥物如異戊巴比妥(pKa=7.9)在胃腸道pH範圍內基本都是非離子型,吸收都快而完全。
少數與正常代謝物相似的藥物,如5-氟尿嘧啶、甲基多巴等的吸收是靠細胞中的載體主動轉運(active transport)而吸收的,這一主動轉運機制對藥物在體內分佈及腎排泄關係比較密切。易化擴散(facilitated diffusion)是靠載體順濃度梯度跨膜轉運方式,如葡萄糖的吸收,吸收速度較快。固體藥物不能吸收,片劑、膠囊劑在胃腸道必須先崩解(disintegration)、溶解(dissolution)後纔可能被吸收。
1.胃腸道給藥 口服(per os)給藥是最常用的給藥途徑。小腸內pH接近中性,粘膜吸收面廣,緩慢蠕動增加藥物與粘膜接觸機會,是主要吸收部位。藥物吸收後通過門靜脈進入肝臟。有些藥物首次通過肝臟就發生轉化,減少進入體循環量,叫做首關消除(first pass elimination)。多數藥物口服雖然方便有效,但其缺點是吸收較慢,欠完全,不適用於在胃腸破壞的,對胃刺激大的,首關消除多的藥物,也不適用於昏迷及嬰兒等不能口服的病人。舌下(sublingual)及直腸(per rectum)給藥雖可避免首關消除,吸收也較迅速,但吸收不規則,較少應用。
2.注射給藥 靜脈注射(intravenous,iv)可使藥物迅速而準確地進入體循環,沒有吸收過程。肌肉注射(intramuscular,im)及皮下注射(subcutaneous,sc)藥物也可全部吸收,一般較口服快。吸收速度取決於局部循環,局部熱敷或按摩可加速吸收,注射液中加入少量縮血管藥則可延長藥物的局部作用。動脈注射(intra-arterial,ia)可將藥物輸送至該動脈分佈部位發揮局部療效以減少全身反應。例如將溶纖藥直接用導管注入冠狀動脈以治療心肌梗塞。注射給藥還可將藥物注射至身體任何部位發揮作用,如局部麻醉。注射給藥需要醫護進行,不方便,如果計算劑量有誤,過量注入將無法回收。
3.呼吸道給藥 肺泡表面積大(達200m2),與血液只隔肺泡上皮及毛細管內皮各一層,而且血流量大,藥物只要能到達肺泡,吸收極其迅速,氣體及揮發性藥物(如全身麻醉藥)可直接進入肺泡。藥物溶液需要經噴霧器分散爲微粒,氣霧劑(aerosol)可將藥液霧化爲直徑達5μm左右微粒,可以達到肺泡而迅速吸收,如在霧化器及口鼻罩間加用一個氣室則效果更好。2~5μm直徑以下的微粒可重被呼出,10μm直徑微粒可在小支氣管沉積。後者可用於異丙腎上腺素治療支氣管哮喘。較大霧粒的噴霧劑(nebula)只能用於鼻咽部的局部治療,如抗菌、消炎、祛痰、通鼻塞等。
4.經皮(transdermal)給藥 除汗腺外,皮膚不透水,但脂溶性藥物可以緩慢通透。許多殺蟲藥可以經皮吸收中毒。利用這一原理可以經皮給藥以達到局部或全身藥效,近年來有許多促皮吸收劑加氮酮(azone),可與藥物製成貼皮劑,如硝苯地平貼皮劑以達到持久的全身療效,對於容易經皮吸收的硝酸甘油也可製成緩釋貼皮劑預防心絞痛發作,每日只貼一次。
4.2 分佈
藥物進入循環後首先與血漿蛋白結合(plasma protein binding)。酸性藥物多與清蛋白結合,鹼性藥物多與α1酸性糖蛋白結合,還有少數藥物與球蛋白結合。這種結合和藥物與受體蛋白結合情況相似:
可見藥物的血漿蛋白結合量([DP])受藥物濃度([D]),血漿蛋白(P)的質和量及解離常數(KD)影響,各藥不同而且結合率(血中與蛋白結合的藥物與總藥量的比值)隨劑量增大而減少。藥理學書籍收載藥物的血漿蛋白結合率是在常用劑量範圍內對正常人測定的數值。藥物與血漿蛋白的結合是可逆性的,結合後藥理活性暫時消失,結合物分子變大不能通過毛細管壁暫時“儲存”於血液中。上述反應式中縱向虛線代表毛細管壁,在吸收過程中游離藥物穿透毛細管壁進血液後與血漿蛋白結合,反應平衡向右移,有利於吸收。在消除過程中(如肝攝取及腎小管分泌),血中游離藥物被除去,反應平衡左移,有利於消除。藥物與血漿蛋白結合特異性低,而血漿蛋白結合點有限,兩個藥物可能競爭與同一蛋白結合而發生置換現象。如某藥結合率達99%,當被另藥置換而下降1%時,則遊離型(具有藥理活性)藥物濃度在理論上將增加100%,可能導致中毒。但一般藥物在被置換過程中,遊離型藥物會加速被消除,血漿中游離型藥物濃度難以持續增高。藥物也可能與內源性代謝物競爭與血漿蛋白結合,例如磺胺藥置換膽紅素與血漿蛋白結合,在新生兒可能導致核黃疸症。血漿蛋白過少(如肝硬化)或變質(如尿毒症)時藥物血漿蛋白結合率下降,也容易發生毒性反應。
吸收的藥物通過循環迅速向全身組織輸送,首先向血流量大的器官分佈(distribution),然後向血流量小的組織轉移,這種現象稱爲再分佈(redistribution),如硫噴妥先在血流量大的腦中發揮麻醉效應,然後向脂肪等組織轉移,效應很快消失。經過一段時間後血藥濃度趨向“穩定”,分佈達到“平衡”,但各組織中藥物並不均等,血漿藥物濃度與組織內濃度也不相等。這是由於藥物與組織蛋白親和力不同所致。因此這種“平衡”稱爲假平衡(pseudoequilibrium),這時血漿藥物濃度高低可以反映靶器官藥物結合量多少。藥物在靶器官濃度決定藥物效應強弱,故測定血漿藥物濃度可以估算藥物效應強度。某些藥物可以分佈至脂肪、骨質等無生理活性組織形成儲庫,或結合於毛髮指(趾)甲組織。藥物的pKa及體液pH是決定藥物分佈的另一因素,細胞內液pH(約爲7.0)略低於細胞外液(約7.4),弱鹼性藥物在細胞內濃度略高,弱酸性藥物在細胞外液濃度略高,根據這一原理,弱酸性藥物苯巴比妥中毒時用碳酸氫鈉鹼化血液及尿液可使腦細胞中藥物向血漿轉移並加速自尿排泄,是重要救治措施之一。
血腦屏障(blood-brain barrier)腦是血流量較大的器官,但藥物在腦組織濃度一般較低,這是由於血腦屏障所致。在組織學上血腦屏障是由血-腦、血-腦脊液及腦脊液-腦三種屏障的總稱,實際上能阻礙藥物穿透的主要是前二者。腦毛細血管內皮細胞間緊密聯接,基底膜外還有一層星狀細胞包圍,藥物較難穿透。腦脊液不含蛋白質,即使少量未與血漿蛋白結合的脂溶性藥物可以穿透進入腦脊液,其後藥物進入靜脈的速度較快,故腦脊液中藥物濃度總是低於血漿濃度,這是大腦自我保護機制。治療腦病可以選用極性低的脂溶性藥物,例如磺胺藥中的磺胺嘧啶。爲了減少中樞神經不良反應,對於生物鹼可將之季銨化以增加其極性,例如將阿托品季銨化變爲甲基阿托品後不能通過血腦屏障,即不致發生中樞興奮反應。
胎盤屏障(placenta barrier)是胎盤絨毛與子宮血竇間的屏障,由於母親與胎兒間交換營養成分與代謝廢物的需要,其通透性與一般毛細管無顯著差別,只是到達胎盤的母體血流量少,進入胎兒循環慢一些罷了。例如母親注射磺胺嘧啶2小時後才能與胎兒達到平衡。利用這一原理可以在預期胎兒娩出前短時內注射鎮靜鎮痛藥,新生兒不致遭受影響。應該注意的是幾乎所有藥物都能穿透胎盤屏障進入胚胎循環,在妊娠期間應禁用對胎兒發育有影響的藥物。
4.3 生物轉化
藥物,作爲外來活性物質(xenobiotic),機體首先要將之滅活,同時還要促其自體內消除。能大量吸收進入體內的藥物多是極性低的脂溶性藥物,在排泄過程中易被再吸收,不易消除。體內藥物主要在肝臟生物轉化(biotransformation)而失去藥理活性,並轉化爲極性高的水溶性代謝物而利於排出體外。生物轉化與排泄統稱爲消除(elimination)。
生物轉化分兩步進行,第一步爲氧化、還原或水解,第二步爲結合。第一步反應使多數藥物滅活,但少數例外反而活化,故生物轉化不能稱爲解毒過程。第二步與體內物質結合後總是使藥物活性降低或滅活並使極性增加。各藥在體內轉化過程不同,有的只經一步轉化,有的完全不變自腎排出,有的經多步轉化生成多個代謝產物。
肝臟微粒體的細胞色素P-450酶系統是促進藥物生物轉化的主要酶系統,故又簡稱肝藥酶,現已分離出70餘種。此酶系統的基本作用是從輔酶Ⅱ及細胞色素b5獲得兩個H+,另外接受一個氧分子,其中一個氧原子使藥物羥化,另一個氧原子與兩個H+結合成水(RH+NADPH+O2+2H+→ROH+NADP++H2O),沒有相應的還原產物,故又名單加氧酶,能對數百種藥物起反應(圖3-1)。此酶系統活性有限,在藥物間容易發生競爭性抑制。它又不穩定,個體差異大,且易受藥物的誘導或抑制。例如苯巴比妥能促進光面肌漿網增生,其中P-450酶系統活性增加,加速藥物生物轉化,這是其自身耐受性及與其他藥物交叉耐受性的原因。西米替丁抑制P-450酶系統活性,可使其他藥物效應敏化。該酶系統在缺氧條件下可對偶氮及芳香硝基化合物產生還原反應,生成胺基(圖3-2)。微粒體內還存在水解酶及葡萄糖醛酸轉移酶。
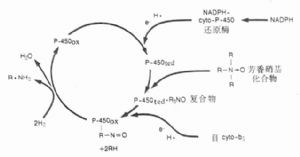

生物轉化的第二步反應是結合。多數經過氧化反應的藥物再經肝微粒體的葡萄糖醛酸轉移酶作用與葡萄糖醛酸結合。有些藥物還能和乙酰基、甘氨酸、硫酸等結合。這些結合反應都需要供體參加,例如二磷酸尿嘧啶是葡萄糖醛酸的供體。藥物在體內轉化過程,舉例說明見表3-1。
表3-1 藥物生物轉化類型舉例
| 轉化類型 | 轉化反應通式 | 酶系 | 藥物舉例 |
| 1.氧化 脂肪族羥化 芳香族羥化 N去烷基 O去烷基 硫氧化 去硫 去滷 環氧化 醇類氧化 醛類氧化 胺類氧化 嘌呤氧化 2.還原 硝基還原 偶氮還原 醛類還原 酮類還原 3.水解 酰胺鍵水解 酯鍵水解 4.結合 葡萄糖醛酸 結合 乙酰化 | R→ROH Ar→ArOH CH3 | R1―N―R2→R1―NH―R2 R―O―CH3→ROH O ‖ R1―S―R2→R1―S―R2S O ‖ ‖ R1―P―R2→R1―P―R2X OH | | R1―CH―R2→R1―CH―R2+HX O / \ R1―CH=CH―R2→R1―CH―CH―R2R―CH2OH→RCHO RCHO→RCOOH RCH2NH2→RCHO+NH2Ar(N)→Ar(O)ArNO2→ArNH2Ar1―N=N―Ar2→Ar1NH2+Ar2NH2RCHO→RCH2OH O OH ‖ | R1―C―R2→R1―CH―R2 R1―CONH―R2→R1COOH+R2NH2R1COOR2→R1COOH+R2OH 載體:UDP-葡萄糖醛酸 載體:乙酰輔酶A | 微粒體酶 微粒體酶 微粒體酶 微粒體酶 微粒體酶 微粒體酶 微粒體酶 微粒體酶 非微粒體酶 非微粒體酶 非微粒體酶 非微粒體酶 微粒體酶 微粒體酶 非微粒體酶 非微粒體酶 微粒體酶 非微粒體酶 微粒體酶 非微粒體酶 | 司可巴比妥 苯妥英 地西泮 可待因 氯丙嗪 對硫磷 氟烷 苯並芘(致癌物) 乙醇 乙醛 腎上腺素,組胺 茶鹼 氯硝西泮 百浪多息 水合氯醛 納洛酮 利多卡因,普魯卡因胺 乙酰膽鹼,普魯卡因 氯黴素,嗎啡 異煙肼 |
4.4 排 泄
藥物在體內最後的過程是排泄(excretion),腎臟是主要排泄器官。遊離的藥物能通過腎小球過濾進入腎小管。隨着原尿水分的回收,藥物濃度上升。當超過血漿濃度時,那些極性低、脂溶性大的藥物反向血漿擴散(再吸收),排泄較少也較慢。只有那些經過生物轉化的極性高、水溶性代謝物不被再吸收而順利排出。有些藥物在近曲小管由載體主動轉運入腎小管,排泄較快。在該處有兩個主動分泌通道,一是弱酸類通道,另一是弱鹼類通道,分別由兩類載體轉運,同類藥物間可能有競爭性抑制。例如丙磺舒抑制青黴素主動分泌,使後者排泄減慢,藥效延長並增強。鹼化尿液使酸性藥物在尿中離子化,酸化尿液使鹼性藥物在尿中離子化,利用離子障原理阻止藥物再吸收,加速其排泄,這是藥物中毒常用的解毒方法(圖3-3)。

藥物可自膽汁排泄,原理與腎排泄相似,但不是藥物排泄的主要途徑。藥物自膽排泄有酸性、鹼性及中性三個主動排泄通道。有些藥物在肝細胞與葡萄糖醛酸等結合後排入膽中,隨膽汁到達小腸後被水解,遊離藥物被重吸收,稱爲肝腸循環(hepato-enteral circulation)。在膽道引流病人,藥物的血漿半衰期將顯著縮短,如氯黴素、洋地黃等。乳汁pH略低於血漿,鹼性藥物可以自乳汁排泄,哺乳嬰兒可能受累。胃液酸度更高,某些生物鹼(如嗎啡等)注射給藥也可向胃液擴散,洗胃是中毒治療和診斷的措施。藥物也可自唾液及汗液排泄。糞中藥物多數是口服未被吸收的藥物。
5 體內藥量變化的時間過程
體內藥量隨時間而變化的過程是藥動學研究的中心問題。藥量與效應的關係(量效關係)已在藥效學章詳述。加入時間因素就引出時量關係(time-concentration relationship)與時效關係(time-response relationship)。大多數情況下由於量效關係基本固定,在達到“平衡”後兩條曲線平行一致。整體動物一次血管外給藥的時量(效)曲線見圖3-4。按一室模型理解,曲線升段主要是吸收過程(此時消除過程已經開始)。曲線在峯值濃度(peak concentration, Cmax)時吸收速度與消除速度相等。從給藥時至峯值濃度的時間稱爲達峯時間(peak time, Tpeak),曲線降段主要是藥物消除過程。血藥濃度下降一半的時間稱爲消除半衰期(elimination half-life time)。血藥濃度超過有效濃度(低於中毒濃度)的時間稱爲有效期(effective peroid)。曲線下面積(area under the curve, AUC)與吸收入體循環的藥量成比例,反映進入體循環藥物的相對量。AUC是血藥濃度(C)隨時間(t)變化的積分值:

當t1爲0,t2爲∞時,AUC=Co/Ke,單位是g.h.L-1。
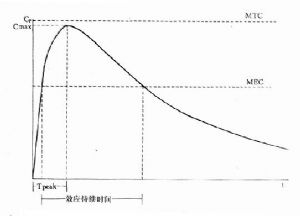
圖3-4 ,典型時量曲線圖
MTC最小中毒濃度 MEC最小有效濃度

F(AUC)相等,但Tpeak及 Cmax不等
生物利用度(bioavailability)是指經過肝臟首關消除過程後能被吸收進入體循環的藥

劑口服後測得的量效曲線,其AUC相等(表示F值相等),但Tpeak及Cmax不等,吸收快的Cmax可能已超過最低中毒濃度,吸收慢的Cmax可能還在有效濃度以下。生物利用度是藥物製劑質量的一個重要指標。
6 藥物消除動力學
從生理學看,體液被分爲血漿、細胞間液及細胞內液幾個部分。爲了說明藥動學基本概念及規律現假定機體爲一個整體,體液存在於單一空間,藥物分佈瞬時達到平衡(一室模型)。問題雖然被簡單化,但所得理論公式不失爲臨牀應用提供了基本規律。按此假設條件,藥物在體內隨時間變化可用下列基本通式表達:dC/dt=kCn。C爲血藥濃度,常用血漿藥物濃度。k爲常數,t爲時間。由於C爲單位血漿容積中的藥量(A),故C也可用A代替:dA/dt=kCn,式中n=0時爲零級動力學(zero-order kinetics),n=1時爲一級動力學(first-order kinetics),藥物吸收時C(或A)爲正值,消除時C(或A)爲負值。在臨牀應用中藥物消除動力學公式比較常用,故以此爲例如以推導和說明。
6.1 零級消除動力學
當n=0時,-dC/dt=KC0=K(爲了和一級動力學中消除速率常數區別,用K代k),將上式積分得:
Ct=C0- Kt,C0爲初始血藥濃度,Ct爲t時的血藥濃度,以C爲縱座標、t爲橫座標作圖呈直線(圖3-6),斜率爲K,當Ct/C0=1/2時,即體內血漿濃度下降一半(或體內藥量減少一半)時,t爲藥物消除半衰期(half-life time, t1/2)。
按公式1/2C0=C0-Kt1/2

可見按零級動力學消除的藥物血漿半衰期隨C0下降而縮短,不是固定數值。零級動力學公式與酶學中的Michaelis-Menten公式相似: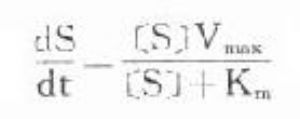 ,式中S爲酶的底物,Vmax爲最大催化速度,Km爲米氏常數。當[S]>>Km時,Km可略去不計,ds/dt=Vmax,即酶以其最大速度催化。零級動力學公式與此一致,說明當體內藥物過多時,機體只能以最大能力將體內藥物消除。消除速度與C0高低無關,因此是恆速消除。例如飲酒過量時,一般常人只能以每小時10ml乙醇恆速消除。當血藥濃度下降至最大消除能力以下時,則按一級動力學消除。
,式中S爲酶的底物,Vmax爲最大催化速度,Km爲米氏常數。當[S]>>Km時,Km可略去不計,ds/dt=Vmax,即酶以其最大速度催化。零級動力學公式與此一致,說明當體內藥物過多時,機體只能以最大能力將體內藥物消除。消除速度與C0高低無關,因此是恆速消除。例如飲酒過量時,一般常人只能以每小時10ml乙醇恆速消除。當血藥濃度下降至最大消除能力以下時,則按一級動力學消除。

體內藥物過多,超過機體最大消除能力(虛線)時爲零級動力學恆速消除
體內藥物降至虛線以下時爲一級動力學恆比消除。插圖縱座標爲對數標尺
6.2 一級消除動力學
當n=1時,-dC/dt=keC1=keC,式中k用ke表示消除速率常數 (elimination rate constant)。將上式積分得

可見按一級動力學消除的藥物半衰期與C高低無關,是恆定值。體內藥物按瞬時血藥濃度(或體內藥量)以恆定的百分比消除,單位時間內實際消除的藥量隨時間遞減。消除速率常數(ke)的單位是h-1,它不表示單位時間內消除的實際藥量,而是體內藥物瞬時消除的百分率。例如ke=0.5h-1不是說每小時消除50%(如果t1/2=1小時則表示每小時消除50%)。按t1/2=0.693/ke計算t1/2=1.39h,即需1.39h後才消除50%。再按 計算,1小時後體內尚存60.7%。絕大多數藥物都按一級動力學消除。這些藥物在體內經過t時後尚存

當n=5時,At≈3%A0,即經過5個t1/2後體內藥物已基本消除乾淨。與此相似,如果每隔一個t1/2給藥一次(A0),則體內藥量(或血藥濃度)逐漸累積,經過5個t1/2後,消除速度與給藥速度相等,達到穩態(steady state):
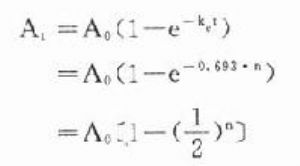
當n=5時,At≈97%A0。這一時間,即5個t1/2不因給藥劑量多少而改變。具體數值見表3-2。
藥物自體內消除的一個重要指標是血漿清除率(plasma clearance,Cl),是肝腎等的藥物消除率的總和,即單位時間內多少容積血漿中的藥物被消除乾淨,單位用L·h-1(也有人用ml·min-1,和肌酐消除率一致)或按體重計算 L·kg-1·h-1。按定義,CL=RE/Cp,RE是消除速率(rate of elimination),即單位時間內被機體消除的藥量,Cp爲當時的血漿藥物濃度。由於RE非固定值也不易檢測,故常用表觀分佈容積(apparent volume of distribution, Vd)計算。 Vd是指靜脈注射一定量(A)藥物待分佈平衡後,按測得的血漿濃度計算該藥應占有的血漿容積。事實上靜注藥物後未待分佈平衡已有部分藥物自尿排泄及(或)在肝轉化而消除,故必需多次檢測Cp,作時量曲線圖,將穩定下降的消除段向O時延升至和Y軸交點以求得理論上靜注藥量A在體內分佈平衡時的血漿濃度C0,以此算出Vd=A/C0(圖3-7)。按RE=keA,Cp=A/Vd,故Cl=keVd。在一級動力學的藥物中,Vd及Cl是兩個獨立的藥動學指標,各有其固定的數值,互不影響,也不因劑量大小而改變其數值。Vd是表觀數值,不是實際的體液間隔大小。除少數不能透出血管的大分子藥物外,多數藥物的Vd值均大於血漿容積。與組織親和力大的脂溶性藥物其Vd可能比實際體重的容積還大。Cl也不是藥物的實際排泄量。它反映肝和(或)腎功能,在肝和(腎)功能不足時Cl值會下降,因爲Cl是肝腎等消除能力的總和。肝清除率雖然難測,但有重要的理論意義。肝清除率小的藥物,首關消除少,其口服生物利用度大,但易受肝功能,血漿蛋白結合力及肝藥酶誘導或抑制藥的影響。肝清除率大的藥物,首關消除多,其口服生物利用度小。有些藥物的肝清除率很高,接近肝血流量,稱爲灌流限制性清除,其肝清除率受肝血流量影響較大。藥物以原形自腎消除的百分率比較容易測定。自腎排泄多的藥物易受腎功能影響,自腎排泄少的藥物易受肝功能影響。醫生可以據此在肝或腎功能不足病人適當調整劑量。在零級動力學的藥物中,RE以恆速消除,不隨Cp下降而改變,故Cl 值不固定,與Cp成反比。
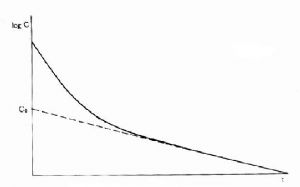
圖3-7 表觀分佈容積計算法
C0是靜注藥量A在0時理論上的血藥濃度
Cl值實際上常用靜脈或肌肉注射藥物A後測定Cp,繪出時量曲線,算出AUC再按CL=A/AUC取得。因爲AUC=C0/ke,代入得
CL=keVd=C0Vd/AUC=A/AUC。
6.3 連續恆速給藥
臨牀治療常需連續給藥以維持有效血藥濃度。在一級動力學藥物中,開始恆速給藥時藥物吸收快於藥物消除,體內藥物蓄積。按 計算約需5個t1/2達到血藥穩態濃度(Css)(圖3-8),此時給藥速度(RA)與消除速度(RE)相等。
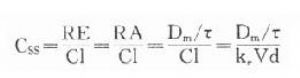 (τ爲給藥間隔時間)可見Css隨給藥速度(RA=Dm/τ)快慢而升降,到達Css時間不因給藥速度加快而提前,它取決於藥物的ke或t1/2。據此,可以用藥物的keVd或Cl計算給藥速度以達到所需的有效藥物濃度。靜脈恆速滴注時血藥濃度可以平穩地到達Css。分次給藥雖然平均血藥濃度上升與靜脈滴注相同,但實際上血藥濃度上下波動(圖3-8)。分藥間隔時間越長波動越大,其峯值濃度
(τ爲給藥間隔時間)可見Css隨給藥速度(RA=Dm/τ)快慢而升降,到達Css時間不因給藥速度加快而提前,它取決於藥物的ke或t1/2。據此,可以用藥物的keVd或Cl計算給藥速度以達到所需的有效藥物濃度。靜脈恆速滴注時血藥濃度可以平穩地到達Css。分次給藥雖然平均血藥濃度上升與靜脈滴注相同,但實際上血藥濃度上下波動(圖3-8)。分藥間隔時間越長波動越大,其峯值濃度 ,谷值濃度 Css-min=Css- maxe 。如果實際Css過高或過低,可以按已達到的Css與需要達到的Css比值調整給藥速度,即Css(已達到的)/Css(需要的)=RA(現用的)/RA(將調整的)
,谷值濃度 Css-min=Css- maxe 。如果實際Css過高或過低,可以按已達到的Css與需要達到的Css比值調整給藥速度,即Css(已達到的)/Css(需要的)=RA(現用的)/RA(將調整的)

圖3-8 連續恆速給藥時的時量曲線
血藥濃度波動越小。給藥劑量越大,血藥濃度越高
但從調整劑量時開始需再經過5個t1/2方能達到需要的Css。
在病情危重需要立即達到有效血藥濃度時,可於開始給藥時採用負荷劑量(loading dose,D1),因爲

Ass就是負荷劑量。可將第一個t1/2內靜脈滴注量的1.44倍在靜脈滴注開始時推注入靜脈即可立即達到並維持Css。在分次恆速給藥達到Css時,體內Ass是維持劑量(maintenance dose, Dm)與體內上一劑量殘留藥物的和,即

當給藥間隔時間τ=t1/2時,

即每隔一個t1/2給藥一次時採用首劑加倍劑量的D1可使血藥濃度迅速達到Css。
理想的給藥方案應該是使CSS- max略小於最小中毒血漿濃度(MTC)而CSS-min略大於最小有效血漿濃度(MEC),即血藥濃度波動於MTC與MEC之間治療窗,這一Dm可按下列公式計算:
Dm=(MTC - MEC)Vd
負荷劑量計算法與上同,即D1=ASS=1.44t1/2 RA=1.44t1/2 Dm/τ,τ爲給藥間隔時間。τ可按一級消除動力學公式推算得
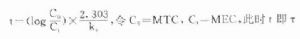

因此可以根據藥物的MTC及MEC利用這些公式計算出D1,Dm及τ。注意此時τ≠t1/2,D1≠2Dm(圖3-9)。

在零級動力學藥物中,體內藥量超過機體最大消除能力。如果連續恆速給藥,RA>RE,體內藥量蓄積,血藥濃度將無限增高。停藥後消除時間也較長,超過5個t1/2。因爲t1/2=0.5C0/K,達到C0越高t1/2越長。
臨牀用藥可根據藥動學參數如Vd、Cl、ke、t1/2及AUC等按以上各公式計算劑量及設計給藥方案以達到並維持有效血藥濃度。除了少數t1/2特長或特短的藥物,或零級動力學藥物外,一般可採用每一個半衰期給於半個有效量(half dose at half life interval)並將首次劑量加倍是有效、安全、快速的給藥方法。
有些藥在體內轉化爲活性產物則需注意此活性產物的藥動學,如果活性產物的消除是藥物消除的限速步驟的話,則應按該產物的藥動學參數計算劑量及設計給藥方案。
6.4 一級藥動學指標間的相互關係
1.F=A/D×100%口服劑量(D)由於不能100%吸收及存在首關消除效應,能進入體循環的藥量(A)只佔D的一部分,這就是生物利用度(F)。藥動學計算時應採用絕對生物利用度,相對生物利用度作爲評比藥物製劑質量的指標。生物利用度還包括吸收速度問題,達峯時間(Tpeak)是一個參考指標。
2.A=C·Vd或C=A/Vd體內藥量(A)與血藥濃度(C)比值固定,在許多藥動學公式中,A與C可以通用,如At=也可用Ct=。
3.Cp=[D]+[DP] 血漿中藥物有遊離型(D)與血漿蛋白結合型(DP),定量測定時需將血漿蛋白沉澱除去,故通常所說的血漿藥物濃度(Cp)是指[D]與[DP]的總和。只有透析法或超離心法纔可能將二者分離以計算藥物的血漿蛋白結合率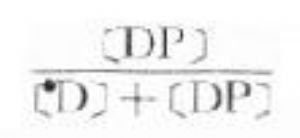 ×100% 。
×100% 。
4. 曲線下面積(AUC)是一個可用實驗方法測定的藥動學指標。它反映進入體循環藥量的多少。時量曲線某一時間區段下的AUC反映該時間內的體內藥量。AUC是獨立於房室模型的藥動學參數,常用於估算血漿清除率(Cl)。
曲線下面積(AUC)是一個可用實驗方法測定的藥動學指標。它反映進入體循環藥量的多少。時量曲線某一時間區段下的AUC反映該時間內的體內藥量。AUC是獨立於房室模型的藥動學參數,常用於估算血漿清除率(Cl)。
5.ke=0.693/t1/2=RE/A=CL/Vd 消除速率常數是藥物瞬時消除的百分率而不是單位時間藥物消除速率(RE),是決定t1/2的參數,但其本身又取決於Cl及Vd,故不是獨立的藥動學指標。
6.Vd=A/C0=A/AUC ke 表現分佈容積(Vd)是獨立的藥動學指標,不是實際的體液容積,取決於藥物在體液的分佈。Vd大的藥物與組織蛋白結合多,主要分佈於細胞內液及組織間液。Vd小的藥物與血漿蛋白結合多,較集中於血漿。Vd不因A多少而變化。
7.CL=keVd=RE/Cp=A/AUC 血漿清除率(Cl)是肝腎等清除率的總和,也不是實際的藥物消除速率(RE),是另一個獨立於A的重要藥動學指標,但受肝腎功能的影響。
8.t1/2=0.693/ke=0.693Vd/CL 血漿藥物消除半衰期(t1/2)是一個非常實用的藥動學指標,雖然獨立於A,但受Cl及Vd雙重製約,Cl大時t1/2短,Vd大時t1/2長。例如慶大黴素Cl小(60ml·min-1),Vd也小(0.25L·kg-1),其t1/2不長(2~3h)。氯喹Cl大(700ml·min-1),Vd也大(185L·kg-1),其t1/2並不短(8天)。藥物在吸收及分佈過程中也有半衰期,分別用t1/2a及t1/2α表示。
9.穩態時RA=RE=CSS·Cl=CSS·Vd·ke
故 CSS是恆速連續給藥達到穩態時平均血藥濃度,應該和預期的有效濃度相等。必要時可以按達到的CSS與預期的CSS比值調整劑量或給藥速度(RA)。
CSS是恆速連續給藥達到穩態時平均血藥濃度,應該和預期的有效濃度相等。必要時可以按達到的CSS與預期的CSS比值調整劑量或給藥速度(RA)。
10.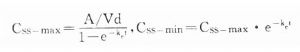 分次定時定量給藥時,CSS上下波動。當每t1/2給藥一次時,其峯值(CSS- max)與谷值(CSS- min)的比值爲2,縮短給藥間隔可以減少CSS波動。
分次定時定量給藥時,CSS上下波動。當每t1/2給藥一次時,其峯值(CSS- max)與谷值(CSS- min)的比值爲2,縮短給藥間隔可以減少CSS波動。
11. 每t1/2給藥一次時,首次給予加倍劑量,即負荷劑量(D1)可以立即達到CSS。
每t1/2給藥一次時,首次給予加倍劑量,即負荷劑量(D1)可以立即達到CSS。
6.5 房室模型
以上所述各種藥動學公式都是將機體視爲一個整體空間,假設藥物在其中轉運迅速,瞬時達到分佈平衡的條件下推導而得的。實際上機體絕非如此簡單,不僅有血漿、細胞外液及細胞內液等間隔,而且各組織細胞間存在着無數的區間。靜脈注射藥物的時量(對數標尺)關係並非直線,而是一條由無數區段組成的連續弧線。粗略地看可見早期一段快速下降,後來才逐漸穩定緩慢下降。這是因爲藥物進入血液循環後快速向組織分佈,首先進入血注量大的肺、腎、心、腦等器官,然後再向其他組織分佈,最後達到平衡(假平衡)。因此設想機體由幾個互相連通的房室(compartment)組成。這個房室不是解剖學上分隔體液的房室,而是按藥物分佈速度以數學方法劃分的藥動學概念。多數藥物按二房室模型轉運(少數單房室或多房室),中央室大致包括血漿及那些血流量多的器官,周邊室包括機體其餘部分,界限並不明確。時量曲線因此也只能大致分爲分佈相及消除相兩個指數衰減區段(圖3-10)。其藥動學規律與單房室不同,如C=Ae-αt +Be-βt,α及β分別爲分佈相(A)及消除相(B)的消除速率常數。而且在分佈相中Vd逐漸增大,ke(α)逐漸減少,t1/2逐漸延長,因此藥動學計算需要特殊處理。即使在消除相,血藥濃度穩定線性下降,各組織濃度及其下降速度也不盡相等,故稱假平衡。可見問題非常複雜。
圖3-10 二房室模型時量曲線
B.消除相(實線)與消除曲線(虛線)
正由於問題過於複雜,臨牀應用諸多不便,實際運算也存在諸多困難。房室模型並非藥物固有的藥動學指標,機體也無此解剖學間隔,即使運用電子計算機擬合也不一定獲得明確的劃分。用同一藥物試驗,在某些人呈二室模型,另些人可能呈一室或三室模型。同一藥物靜脈注射時呈二室模型而口服則呈單一房室模型。在分佈相時藥物實際上已開始消除,到達消除相時可能已有相當分量的藥物已被消除。如果用血管外給藥(口服、肌注等)分佈相常被吸收相掩蓋。這些時相的劃分僅靠血藥濃度的測定。如果早期(此時血藥濃度變化較快)取樣間隔過疏,很難據此準確劃分時相,因此,越來越多的臨牀家及研究者逐漸放棄房室模型而轉向採用適用於所有藥物的無房室方法(noncompartmental medtod)來解決實際問題,對此,有待今後深入學習。
從另一方面看,時量曲線在達到假平衡後已呈單一指數衰減的直線,此時房室劃分已無需要,可以按β值計算t1/2及其他實用的藥動學指標。
AUC是與房室無關的藥動學指標,可用實驗方法測定。AUC(0-∞)=C0/ke,或AUC(T-∞)=CT/β,T是消除相開始的時間。再用AUC算出Vd及Cl:
Vd=A/AUCβ,CL=A/AUC