3 概述
巴特綜合徵是因腎小球球旁細胞增生,分泌大量的腎素引起的繼發性醛固酮增多症候羣。爲一常染色體隱性遺傳病,由Bartter(1962)首次報道,故稱爲巴特綜合徵。其臨牀特徵爲嚴重的低鉀血癥和代謝性鹼中毒,伴有高腎素、高醛固酮血癥、腎小球旁器增生和肥大及腎小管保鈉和濃縮功能障礙,但無高血壓及水腫且對外源性血管緊張素Ⅱ無反應。臨牀表現主要爲肌無力,週期性麻痹,心律失常,腸麻痹等低鉀症狀及煩渴,夜尿增多,骨質疏鬆等。現認爲本綜合徵是由離子通道基因突變引起的臨牀綜合徵。本病又稱爲先天性醛固酮增多症、慢性特發性低鉀血癥、腎小球旁器增生綜合徵。近年來,分子診斷學研究揭示Bartter綜合徵有3種不同的臨牀和遺傳類型,即先天性Bartter綜合徵,典型Bartter綜合徵和Gitelman綜合徵。通常所說的Bartter綜合徵是指典型Bartter綜合徵。先天性Bartter綜合徵病人發現有兩種基因型,Ⅰ型是由於N-K-2CL-發生失功能性基因突變所致,Ⅱ型是由於ROMK基因突變所致。典型Bartter綜合徵是由於CLC-kb通道基因突變所致。
9 流行病學
巴特綜合徵是一常染色體隱性遺傳病,男性外顯率較高。1962年Bartter首次報告2例,以後陸續有類似報告。本病較少見,迄今報告共200多例,國內僅報道幾十例。估計發病率爲19/100萬。世界各地及所有種族均有報告,但黑人發病率偏高,女性稍多於男性。明確診斷年齡最早爲孕20周,最晚至50歲。本病常見於兒童,5歲之前出現症狀者佔半數以上。本病發病有明顯的家族傾向,但罕見垂直遺傳,遺傳方式符合常染色體隱性遺傳。
10 病因
巴特綜合徵病因尚無定論。多數學者認爲是常染色體隱性遺傳性疾病。曾有一家9個同胞中5個患病和一家連續2代4例患病的報告。現代分子生物學技術也揭示Bartter綜合徵是由腎小管上皮細胞上的離子轉運蛋白基因突變所引起。目前已發現嬰兒型Batter綜合徵存在Na-K-2Cl-基因突變,該基因位於15q12-21,有16個外顯子,編碼1099個氨基酸,爲Na-K-2Cl-通道,已發現20多種突變。經典型Bartter綜合徵系由CICNKB基因突變所致,該基因位於1q38,編碼含687個氨基酸的細胞基底側的Cl-通道,現已發現約20種突變類型。成人型Bartter綜合徵又稱Batter-Gietlman綜合徵,系由噻嗪敏感的Na-K通道基因(SCI12A3)突變所致,該基因定位於16q913,編碼1021個氨基酸,已發現多達40種突變。此外還有一些病人中發現鉀通道基因(ROWK)突變。因此Batter綜合徵可以認定爲由上述幾種離子通道基因突變引起的臨牀綜合徵。
11 發病機制
巴特綜合徵的發病機制尚未完全闡明。有人就本綜合徵發病環節提出4種假說:
1.血管壁對ATI的反應有缺陷導致腎素生成增多和繼發性醛固酮增多。
2.近端小管鈉重吸收障礙導致鈉負平衡;低鈉飲食亦不能逆轉腎性失鉀。
3.前列腺素生成過多,使腎小管失鈉,血鈉減低從而激活腎素-血管緊張素系統。
4.髓襻升支厚壁段對氯化物轉輸障礙,使氯化物重吸收減少,鉀排泄增多導致低鉀血癥;低鉀血癥刺激前列腺素E2的生成,並使血漿腎素活腎素活性和血管緊張素Ⅰ升高。前列腺素E2升高後血管對ATI不敏感,因而血壓正常。
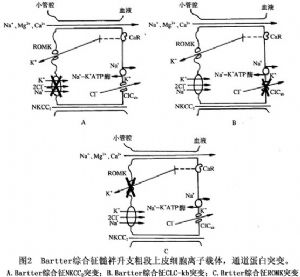
近年來的臨牀與實驗研究對Bartter綜合徵發病機制的認識有了很大的進展,認爲Bartter綜合徵是由於髓襻升支厚壁段穿上皮細胞Cl-、Na的轉運障礙所致。目前對髓襻升支的幾種離子通道蛋白的基因編碼已經克隆出來,由於這些離子通道蛋白髮生了喪失功能的基因突變,致使離子轉運功能發生障礙。正常腎單位髓襻升支厚壁段(圖1)對Cl-、Na再吸收是由對布美他尼敏感的鈉-鉀-2氯運載體(bumetanide-sensitive sodium-potassium-2-chloride transporter,NKCC2)進行的。由於細胞內Na與C1-較細胞外低,NKCC2將Na、K、2Cl-運轉入細胞內,仍維持電中性。上皮細胞的基側膜上有Na-K-ATP酶能把過多的Na泵出細胞外,進入血液。另外,還有腎臟特異性基側氯通道(kidney specific base lateral channel,CIC-kb)把Cl-泵出細胞外,經血液再吸收。髓襻升支厚壁段上的管腔膜上還有ATP調節鉀通道(ATP-regulated potassium channel,ROMK)。NKCC2的轉運速率是由ROMK對鉀再循環進行調節,即ROMK爲NKCC2提供有效的K濃度,保證管腔的正電位。

基因研究推斷,上述離子運載體蛋白或通道蛋白中任何一種發生突變,都可能出現離子轉運障礙,從而導致Bartter綜合徵的發生。不同的通道蛋白或載體的缺陷可形成Bartter綜合徵的不同的亞型。目前認爲,由於NKCC2功能喪失性突變,導致Na、K的再吸收障礙;ClCkb通道蛋白失活,限制了NKCC2運載體的轉運速率,損害了K的再循環過程對K的再吸收。所以,只要上述環節中任何一種環節上發生了功能喪失性突變,都會削減上皮細胞電位差,減少上述離子重吸收的驅動力(圖2)。
髓襻升支厚段再吸收Na、Cl-減少,細胞外液量輕度降低,繼發高腎素、高醛固酮血癥和腎小球旁器增生與肥大。由於氯化鈉大量流經集合管,刺激泌H、泌K,加上高醛固酮血癥,因而引起低鉀血癥和代謝性鹼中毒。腎素-血管緊張素-醛固酮系統功能亢進,促進激肽、血管舒緩素生成,前列腺素生成增多,使血管對血管緊張素反應降低,血壓保持正常,無水腫表現。最近研究發現,Bartter綜合徵患者單核細胞NO合成酶(ecNOS)mRNA水平呈高表達,尿中NO代謝產物NO2-/NO3-與cGMP平行升高,推測由於NO產生增多,減少血管張力,認爲也是Bartter綜合徵患者血管對血管緊張素反應性降低的原因之一。有關ecNOS在Bartter綜合徵發病機制中的作用,尚需深入研究。
12 巴特綜合徵的臨牀表現
巴特綜合徵臨牀表現呈多樣化,臨牀類型不一,主要爲肌無力、週期性麻痹、心律失常、腸麻痹等低鉀症狀及煩渴、夜尿增多、骨質疏鬆等。兒童發病率較高,生長發育延遲,智力下降。目前還沒有其它特殊治療方法。發病以青少年多見,性別無顯著性差異,無種族差異。如果提高對本病的認識,臨牀上並不一定少見,由於合併症及併發症的出現,往往臨牀不易及時準確的診斷。
12.1 水鹽代謝失常型
最多見,突出表現爲低血鉀性鹼中毒。患者來診的主要原因是低血鉀及鹼中毒,其臨牀表現爲:疲乏無力,下肢或周身軟癱呈週期性癱瘓現象;感覺遲鈍,心律失常,腹脹,腸麻痹,腸梗阻,噁心,嘔吐,排尿困難,暈厥,神智障礙,反射遲鈍,腱反射減弱或消失等低血鉀症狀;持續低血鉀可發生糖代謝紊亂,糖耐量減低,胰島素釋放受影響,腦電圖有異常波型。血鉀<3.0mmol/L,尿鉀>50mmol/24h以上。鹼中毒與低血鉀經常同時發生,有手足麻木,抽搐,呼吸氣短,精神興奮或躁動,肌肉顫抖及腹痛等症,Chvostek及Trosseau徵陽性,血pH值>7.45,血漿:HCO3-常>24mEg/L,尿呈鹼性反應。早期病人尿量增多,可達每天5000毫升以上,比重降低,尿滲透壓降低,患者雖有抽搐,但血鈣、磷、AKP,尿鈣均可正常。
由於脫水失鹽,患者經常口乾、口渴、嗜鹽、多飲、多尿、夜尿多、消瘦、體重減輕、便祕、皮膚彈性差、眼窩深陷、眼壓低,脫水較嚴重時尿少,每天僅300~400ml,可發生虛脫、神志障礙或昏迷。血鈉<130mmol/L,血氯<90mmol/L,尿鈉、尿氯排出增加,有效血容量減少,遠曲小管和球旁器進一步發生變化,引起腎素、前列腺素、血管緊張素及醛固酮分泌增多。
Zipser報道兩例本病,其中1例有嚴重低血鎂,作者認爲低血鎂也可興奮腎臟PG增多而引起巴特綜合徵,或是另有原因,需進一步研究。
12.2 以腎臟病爲主要臨牀表現類型
不少見,本病可常有腎盂腎炎,間質性腎炎,失鹽性腎炎,腎小球腎炎合併腎鈣化,腎結石,腎盂積水,腎功能減退等表現。由於慢性腎臟病變遷延不愈,可發生腎性骨病,骨質疏鬆,牙脫落,繼發性甲狀旁腺功能亢進等表現。並可有尿磷增多及糖尿現象。Meget報道一組巴特綜合徵病患者,由於腎功能異常變化而發生尿酸鹽代謝異常,尿酸清除率下降,尿中尿酸鹽排出減少,血液尿酸水平升高,50%患者發生高尿酸血癥,20%患者發生急性痛風性關節性關節炎。正常人痛風病發生率僅爲0.2%~0.3%,而巴特綜合徵病人合併痛風症大大增加,痛風症也可成爲巴特綜合徵的臨牀表現之一。
MeCrldie報道4例本病,其中3例有高尿鈣症。巴特綜合徵合併腎鈣化,腎結石,高尿鈣症並不少見。結石性質可爲草酸鈣、磷酸鈣、尿酸鹽或爲混合性。血清尿酸值>7.0mg/dl者爲高尿酸血癥。尿中尿酸正常值爲0.5~0.8g/24h,正常尿酸清除率爲6~12ml/min,而巴特綜合徵時排出減少。尿鈣值各地區差異較大,一般來說如高於200~250mg/24h,即爲高尿鈣,應尋找尿鈣增高原因。
12.3 血管活性激素平衡失調錶現
巴特綜合徵有高前列腺素,腎素,血管緊張素和醛固酮,其血尿PGA2,PGE,PGF,PGI。都可升高,但主要是PGE升高。PGA2、PGE及PGF增高均可用阿司匹林治療,3個月後恢復正常水平。Bowden報道7例中5例的PGE增高,用吲哚美辛治療後4例PGE下降,排鈉與排鉀減少,血鉀回升,血漿腎素值下降,肌酐清除率降低。巴特綜合徵的尿PGE和血管舒緩素排出量有關,高腎素血癥是繼發於腎臟PG的增加。血管舒緩素-激肽系統和前列腺素-腎素-血管緊張素-醛固酮系統有關。血管舒緩素-激肽系統活性增高,可刺激腎臟合成PGE增多,用吲哚美辛治療後,PGE、血管舒緩素、血漿腎素活腎素活性均可明顯降低,並可使AngⅡ增加敏感性,血鉀恢復正常。本症時AngI也有增高,可達90~200ng/ml,而正常值僅爲50ng/ml以下水平。用吲哚美辛後不能使尿Aldo排出量降低,理由不清。動物實驗證實由腎動脈注入PGE和花生四烯酸後,可增加血漿腎素活腎素活性,用吲哚美辛後可增加AngⅡ的敏感性,也可降低腎素活性。Fujita給本病患者作血管緊張素注入實驗,確實發現對血管緊張素的反應比正常人低下,但應用白蛋白靜脈注射後就會提高反應性,說明血管壁對血管緊張素的抗壓反應是因低鈉、低血容量等而引起。Inada給本病患者每天入鈉175mmol,並以高於20ng/(kg·min)的AngⅡ注入時,舒張壓可升高20mmHg,而收縮壓要大於100ng/(kg·min)的注入速度時,纔有輕微上升,而正常人僅僅在注入20ng/(kg·min)的速度即能提高收縮壓20mmHg,舒張壓20mmHg,顯然巴特徵患者對外源性的AngⅡ反應性不敏感。
巴特綜合徵的PG增高是原發性的,而血漿腎素活腎素活性,血管緊張素及醛固酮增高均爲繼發性的反應。正常血漿Aldo值爲5.0~15.0ng/dl而巴特綜合徵患者可達50ng/dl以上,尿Aldo值正常爲5.0~20.0ng/24h,而巴特綜合徵可達30ng/24h以上或更高。
12.4 其他臨牀表現
兒童時期發病者常有生長發育障礙,生長停滯或緩慢,智力落後及性腺功能低下,但未見垂體侏儒症表現。巴特綜合徵病人腎功能減退時可合併貧血。脫水較嚴重時可伴有血液濃縮,血紅蛋白達16克以上,並伴有紅細胞增多症等。
14 實驗室檢查
1.血鉀、鈉、氯多低於正常水平。
2.血pH值可高於7.46呈鹼血癥,C02CP高於30mmol/L以上。
3.血漿腎素活腎素活性(PRA)增高可達(4.5±2.9)μg/L·h以上。
5.血中PGA、PGE、PGF、PGI均可升高,如PGF可達(138.0±78.0)ng/ml。
6.血管緊張素Ⅱ(AngⅡ)升高可達(95.8±35.2)ng/L。
7.腎功能檢查 BUN可升高達7.0mmol/L以上,毛森試驗比重低,夜尿量增多,並可發現尿中有蛋白及紅白細胞等。
15 輔助檢查
2.心電圖 可發現低血鉀表現。
3.腎圖異常。
4.腎活檢 腎小球旁器增生肥大可在腎活檢時發現,有球旁細胞、緻密斑細胞、極周細胞及球外系膜細胞的數量增多,或細胞呈肥大分泌現象。95%腎素由球旁細胞分泌,1%~5%可由緻密斑細胞、間質細胞或出球小動脈內皮細胞產生腎素。
17 鑑別診斷
1.原發性與繼發性醛固酮增多症 原醛有血壓明顯升高,繼發性醛固酮增多症如肝硬化、心力衰竭、慢性腎炎和妊娠毒血癥則有原發性疾病臨牀表現可資鑑別。另外,原醛還有血漿腎素活腎素活性降低。
2.其他原因引起的週期性癱瘓 如原發性週期性癱瘓、甲亢、Ⅰ型慢性腎小管性酸中毒、棉酚中毒等,這些疾病均無血漿腎素活腎素活性和醛固酮升高。甲亢者有T3和T4升高。
腎小管性酸中毒者有血pH值和CO2結合力降低,棉酚中毒有食用棉子油史等,可與本綜合徵鑑別。
18 巴特綜合徵的治療
巴特綜合徵的治療初期,主要是針對低血鉀及鹼血癥給以對症治療,但療效欠佳。繼而針對高腎素血癥,高醛固酮症給以治療,僅部分有效。近些年來開展對高前列腺素分泌治療,並輔以低血鉀對症治療取得了進展,但仍有報道不完全令人滿意的。
補鉀是必需的措施,但單獨補鉀,血鉀不能恢復至正常水平時,應加用抗醛固酮藥物,如螺內酯(安體舒通)或氨苯蝶啶,可改善療效,一般用量爲60~l80mg/d,大量可引起男性乳房增大,因此,劑量不宜過大。Solomon及Modlinger等用普萘洛爾(心得安)治療本徵,針對高腎素血癥,其療效也不滿意,但普萘洛爾(心得安)合用螺內酯(安體舒通)時可改善療效,且可恢復血鉀水平。Norby等用阿司匹林,每天每公斤體重給予100mg,治療10周後可取得滿意療效,但停藥4天后又回到治療前水平。說明阿司匹林可抑制PG合成,爲有效藥物,副作用小比較安全,可惜作用不持久。以後採用前列腺素合成酶抑制劑吲哚美辛(消炎痛)等藥物治療,均可減少PG合成,降低腎素、血管緊張素及醛固酮的活性,使症狀改善。吲哚美辛(消炎痛)可單用,也可合用螺內酯(安體舒通),但長期應用可有鈉水瀦留,引起水腫或心力衰竭,因此不宜長期大量應用,主張間斷應用。McGredie用二磷酸鹽治療巴特徵的腎鈣化,取得一定療效。如有痛風症可用別嘌醇(吡唑嘧啶醇),或秋水仙鹼等治療,可緩解痛風性關節性關節炎。脫水失鹽患者可補給氯化鈉液,低鎂者可補給鎂鹽治療等。腎功能減退較重者應給予透析治療。缺鈣者應補充鈣劑及活性維生素D劑治療等。