3 PCR引物設計
3.1 PCR引物設計的原則
PCR引物設計的目的是爲了找到一對合適的核苷酸片段,使其能有效地擴增模板DNA序列。因此,引物的優劣直接關係到PCR的特異性與成功與否。
要設計引物首先要找到DNA序列的保守區。同時應預測將要擴增的片段單鏈是否形成二級結構。如這個區域單鏈能形成二級結構,就要避開它。如這一段不能形成二級結構,那就可以在這一區域設計引物。
現在可以在這一保守區域裏設計一對引物。一般引物長度爲15~30鹼基,擴增片段長度爲100~600鹼基對。
讓我們先看看P1引物。一般引物序列中G+C含量一般爲40%~60%。而且四種鹼基的分佈最好隨機。不要有聚嘌呤或聚嘧啶存在。否則P1引物設計的就不合理。應重新尋找區域設計引物。
同時引物之間也不能有互補性,一般一對引物間不應多於4個連續鹼基的互補。
引物確定以後,可以對引物進行必要的修飾,例如可以在引物的5′端加酶切位點序列;標記生物素、熒光素、地高辛等,這對擴增的特異性影響不大。但3′端絕對不能進行任何修飾,因爲引物的延伸是從3′端開始的。這裏還需提醒的是3′端不要終止於密碼子的第3位,因爲密碼子第3位易發生簡併,會影響擴增的特異性與效率。
綜上所述我們可以歸納十條PCR引物的設計原則:
② 產物不能形成二級結構。
③ 引物長度一般在15~30鹼基之間。
④ G+C含量在40%~60%之間。
⑥ 引物自身不能有連續4個鹼基的互補。
⑦ 引物之間不能有連續4個鹼基的互補。
⑧ 引物5′端可以修飾。
⑨ 引物3′端不可修飾。
⑩ 引物3′端要避開密碼子的第3位。
PCR引物設計的目的是找到一對合適的核苷酸片段,使其能有效地擴增模板DNA序列。如前述,引物的優劣直接關係到PCR的特異性與成功與否。對引物的設計不可能有一種包羅萬象的規則確保PCR的成功,但遵循某些原則,則有助於引物的設計。
1.引物的特異性
引物與非特異擴增序列的同源性不要超過70%或有連續8個互補鹼基同源。
2.避開產物的二級結構區
某些引物無效的主要原因是引物重複區DNA二級結構的影響,選擇擴增片段時最好避開二級結構區域。用有關計算機軟件可以預測估計mRNA的穩定二級結構,有助於選擇模板。實驗表明,待擴區域自由能(△G°)小於58.6lkJ/mol時,擴增往往不能成功。若不能避開這一區域時,用7-deaza-2′-脫氧GTP取代dGTP對擴增的成功是有幫助的。
3.長度
寡核苷酸引物長度爲15~30bp,一般爲20~27mer。引物的有效長度:Ln=2(G+C)+(A+T+,Ln值不能大於38,因爲>38時,最適延伸溫度會超過Taq DNA聚合酶的最適溫度(74℃),不能保證產物的特異性。
4.G+C含量
G+C含量一般爲40%~60%。其Tm值是寡核苷酸的解鏈溫度,即在一定鹽濃度條件下,50%寡核苷酸雙鏈解鏈的溫度,有效啓動溫度,一般高於Tm值5~10℃。若按公式Tm=4(G+C)+2(A+T)估計引物的Tm值,則有效引物的Tm爲55~80℃,其Tm值最好接近72℃以使復性條件最佳。
引物中四種鹼基的分佈最好是隨機的,不要有聚嘌呤或聚嘧啶的存在。尤其3′端不應超過3個連續的G或C,因這樣會使引物在G+C富集序列區錯誤引發。
6.引物自身
引物自身不應存在互補序列,否則引物自身會摺疊成髮夾狀結構牙引物本身復性。這種二級結構會因空間位阻而影響引物與模板的復性結合。若用人工判斷,引物自身連續互補鹼基不能大於3bp。
7.引物之間
兩引物之間不應不互補性,尤應避免3′端的互補重疊以防引物二聚體的形成。一對引物間不應多於4個連續鹼基的同源性或互補性。
8.引物的3′端
引物的延伸是從3′端開始的,不能進行任何修飾。3′端也不能有形成任何二級結構可能,除在特殊的PCR(AS-PCR)反應中,引物3′端不能發生錯配。
在標準PCR反應體系中,用2U Taq DNA聚合酶和800μmol/L dNTP(四種dNTP各200μmol/L)以質粒(103拷貝)爲模板,按95℃,25s;55℃,25s;72℃,1min的循環參數擴增HIV-1 gag基因區的條件下,引物3′端錯配對擴增產物的影響是有一定規律的。A∶A錯配使產量下降至1/20,A∶G和C∶C錯七下降至1/100。引物A:模板G與引物G:模板A錯配對PCR影響是等同的。
9.引物的5′端
引物的5′端限定着PCR產物的長度,它對擴增特異性影響不大。因此,可以被修飾而不影響擴增的特異性。引物5′端修飾包括:加酶切位點;標記生物素、熒光、地高辛、Eu3+等;引入蛋白質結合DNA序列;引入突變位點、插入與缺失突變序列和引入一啓動子序列等。
10.密碼子的簡併
如擴增編碼區域,引物3′端不要終止於密碼子的第3位,因密碼子的第3位易發生簡併,會影響擴增特異性與效率。
3.2 常用的PCR引物設計軟件
[1]軟件的引物設計功能主要體現在兩個方面:首先是引物分析評價功能,該功能只有少數商業版軟件能夠做到,其中以“Oligo 6”最優秀;其次是引物的自動搜索功能,各種軟件在這方面的側重點不同,因此自動搜索的結果也不盡相同。
自動搜索功能以“Premier Primer”爲最強且方便使用,“Oligo 6”其次,其他軟件如“Vector NTI Suit”、“Dnasis”、“Omiga”和“Dnastar”都帶有引物自動搜索功能,但搜索結果不是十分理想。要想得到效果很好的引物,在自動搜索的基礎上還要輔以人工分析。筆者認爲引物設計軟件的最佳搭配是“Oligo”和“Premier”軟件合併使用,以“Premier”進行自動搜索,“Oligo”進行分析評價,如此可快速設計出成功率很高的引物。
Premier Primer使用步驟及技巧
“Premier”軟件啓動界面如下
其主要功能在主界面上一目瞭然(按鈕功能如上述)。限制性酶切點分析及基元查找功能比較簡單,點擊該功能按鈕後,選擇相應的限制性內切酶或基元(如-10序列,-35序列等),按確定即可。常見的限制性內切酶和基元一般都可以找到。你還可以編輯或者添加新限制性內切酶或基元。
進行引物設計時,點擊 按鈕,界面如下:
按鈕,界面如下:
進一步點擊 按鈕,出現“search criteria”窗口,有多種參數可以調整。搜索目的(Seach For)有三種選項,PCR引物(PCR Primers),測序引物(Sequencing Primers),雜交探針(Hybridization Probes)。搜索類型(Search Type)可選擇分別或同時查找上、下游引物(Sense/Anti-sense Primer,或Both),或者成對查找(Pairs),或者分別以適合上、下游引物爲主(Compatible with Sense/Anti-sense Primer)。另外還可改變選擇區域(Search Ranges),引物長度(Primer Length),選擇方式(Search Mode),參數選擇(Search Parameters)等等。使用者可根據自己的需要設定各項參數。如果沒有特殊要求,建議使用默認設置。然後按
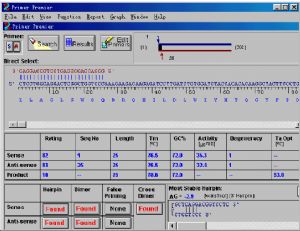
按鈕,出現“search criteria”窗口,有多種參數可以調整。搜索目的(Seach For)有三種選項,PCR引物(PCR Primers),測序引物(Sequencing Primers),雜交探針(Hybridization Probes)。搜索類型(Search Type)可選擇分別或同時查找上、下游引物(Sense/Anti-sense Primer,或Both),或者成對查找(Pairs),或者分別以適合上、下游引物爲主(Compatible with Sense/Anti-sense Primer)。另外還可改變選擇區域(Search Ranges),引物長度(Primer Length),選擇方式(Search Mode),參數選擇(Search Parameters)等等。使用者可根據自己的需要設定各項參數。如果沒有特殊要求,建議使用默認設置。然後按 ,隨之出現的Search Progress窗口中顯示Search Completed時,再按,這時搜索結果以表格的形式出現,有三種顯示方式,上游引物(Sense),下游引物(Anti-sense),成對顯示(Pairs)。默認顯示爲成對方式,並按優劣次序(Rating)排列,滿分爲100,即各指標基本都能達標(如下圖)。
,隨之出現的Search Progress窗口中顯示Search Completed時,再按,這時搜索結果以表格的形式出現,有三種顯示方式,上游引物(Sense),下游引物(Anti-sense),成對顯示(Pairs)。默認顯示爲成對方式,並按優劣次序(Rating)排列,滿分爲100,即各指標基本都能達標(如下圖)。
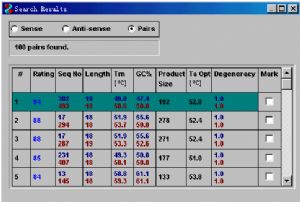
點擊其中一對引物,如第1#引物,並把上述窗口挪開或退出,顯示“Peimer Premier”主窗口,如圖所示:
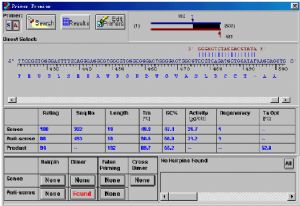
該圖分三部分,最上面是圖示PCR模板及產物位置,中間是所選的上下游引物的一些性質,最下面是四種重要指標的分析,包括髮夾結構(Hairpin),二聚體(Dimer),錯誤引發情況(False Priming),及上下游引物之間二聚體形成情況(Cross Dimer)。當所分析的引物有這四種結構的形成可能時,按鈕由 變成
變成 ,點擊該按鈕,在左下角的窗口中就會出現該結構的形成情況。一對理想的引物應當不存在任何一種上述結構,因此最好的情況是最下面的分析欄沒有,只有。值得注意的是中間一欄的末尾給出該引物的最佳退火溫度,可參考應用。
,點擊該按鈕,在左下角的窗口中就會出現該結構的形成情況。一對理想的引物應當不存在任何一種上述結構,因此最好的情況是最下面的分析欄沒有,只有。值得注意的是中間一欄的末尾給出該引物的最佳退火溫度,可參考應用。
在需要對引物進行修飾編輯時,如在5’端加入酶切位點,可點擊 ,然後修改引物序列。若要回到搜索結果中,則點擊
,然後修改引物序列。若要回到搜索結果中,則點擊 按鈕。
按鈕。
如果要設計簡併引物,只需根據源氨基酸序列的物種來源選擇前述的八種遺傳密碼規則,反推至DNA序列即可。對簡併引物的分析不需像一般引物那樣嚴格。
總之,“Premier”有優秀的引物自動搜索功能,同時可進行部分指標的分析,而且容易使用,是一個相當不錯的軟件。
Oligo 6.22使用技巧簡介
在專門的引物設計軟件中,“Oligo”是最著名的。它的使用並不十分複雜,但初學者容易被其複雜的圖表嚇倒。Oligo 5.0的初始界面是兩個圖:Tm圖和ΔG圖;Oligo 6.22的界面更復雜,出現三個圖,加了個Frq圖。“Oligo”的功能比“Premier”還要單一,就是引物設計。但它的引物分析功能如此強大以至於能風靡全世界。
使用(以Oligo 6.22爲例)
Oligo 6.22的啓動界面如下:

圖中顯示的三個指標分別爲Tm、ΔG和Frq,其中Frq是6.22版本的新功能,爲鄰近6至7個鹼基組成的亞單位在一個指定數據庫文件中的出現頻率。該頻率高則可增加錯誤引發的可能性。因爲分析要涉及多個指標,起動窗口的cascade排列方式不太方便,可從windows菜單改爲tili方式。如果覺得太擁擠,可去掉一個指標,如Frq,這樣界面的結構同於Oligo 5.0,只是顯示更清楚了。
經過Windows/Tili項後的顯示如圖:
在設計時,可依據圖上三種指標的信息選取序列,如果覺得合適,可點擊Tm圖塊上左下角的Upper按鈕 ,選好上游引物,此時該按鈕變成
,選好上游引物,此時該按鈕變成 ,表示上游引物已選取好。
,表示上游引物已選取好。
下游引物的選取步驟基本同上,只是按鈕變成Lower。ΔG值反映了序列與模板的結合強度,最好引物的ΔG值在5’端和中間值比較高,而在3’端相對低(如圖:)

Tm值曲線以選取72℃附近爲佳,5’到3’的下降形狀也有利於引物引發聚合反應。Frq曲線爲“Oligo 6”新引進的一個指標,揭示了序列片段存在的重複機率大小。選取引物時,宜選用3’端Frq值相對較低的片段。
當上下游引物全選好以後,需要對引物進行評價並根據評價對引物進行修改。首先檢查引物二聚體尤其是3’端二聚體形成的可能性。需要注意的是,引物二聚體有可能是上游或下游引物自身形成,也有可能是在上下游引物之間形成(cross dimer)。二聚體形成的能值越高,越不符合要求。一般的檢測(非克隆)性PCR,對引物位置、產物大小要求較低,因而應儘可能選取不形成二聚體或其能值較低的引物。第二項檢查是髮夾結構(hairpin);與二聚體相同,髮夾結構的能值越低越好。一般來說,這兩項結構的能值以不超過4.5爲好。當然,在設計克隆目的的PCR引物時,引物兩端一般都添加酶切位點,必然存在髮夾結構,而且能值不會太低。這種PCR需要通過靈活調控退火溫度以達到最好效果,對引物的髮夾結構的檢測就不應要求太高。第三項檢查爲GC含量,以45-55%爲宜。有一些模板本身的GC含量偏低或偏高,導致引物的GC含量不能被控制在上述範圍內,這時應儘量使上下游引物的GC含量以及Tm值保持接近,以有利於退火溫度的選擇。如果PCR的模板不是基因組DNA,而是一個特定模板序列,那麼最好還進行False priming site的檢測。這項檢查可以看出引物在非目的位點引發PCR反應的可能性。如果引物在錯配位點的引發效率比較高,就可能出假陽性的PCR結果。一般在錯配引發效率以不超過100爲好,但對於特定的模板序列,還應結合比較其在正確位點的引發效率。如果兩者相差很大,比如在正確位點的引發效率爲450以上,而在錯誤位點的引發效率爲130,那麼這對引物也是可以接受的。
當我們結束以上四項檢測,按Alt+P鍵彈出PCR窗口,其中總結性地顯示該引物的位置、產物大小、Tm值等參數,最有用的是還給出了推薦的最佳退火溫度和簡單的評價。
由於“Oligo”軟件的引物自動搜索功能與“Primer Premier 5”的相類似,並且似乎並不比後者更好用,在此不再贅述。其實,使用軟件自動搜索引物就是讓計算機按照人的要求去尋找最佳引物,如果參數設置得當將大大提高工作效率。
除了本地引物設計軟件之外,現在還有一些網上引物設計軟件,如由Whitehead Institute 開發的“Primer 3”等。該軟件的獨特之處在於,對全基因組PCR的引物設計;可以將設計好的引物對後臺核酸數據庫進行比對,發現並排除可引發錯配的引物。因此建議經常做全基因組PCR的用戶試用。
4 PCR反應條件選擇
4.1 PCR反應體系與反應條件
標準的PCR反應體系:
10×擴增緩衝液 10ul
4種dNTP混合物 各200umol/L
引物 各10~100pmol
模板DNA 0.1~2ug
Taq DNA聚合酶 2.5u
Mg2+ 1.5mmol/L
加雙或三蒸水至 100ul
上述體系爲100ul,實驗中一般用50ul體系,即以上所有試劑減半。也可根據Taq酶介紹上的推薦配方。
PCR反應五要素: 參加PCR反應的物質主要有五種即引物、酶、dNTP、模板和Mg2+
引物: 引物是PCR特異性反應的關鍵,PCR 產物的特異性取決於引物與模板DNA互補的程度。理論上,只要知道任何一段模板DNA序列,就能按其設計互補的寡核苷酸鏈做引物,利用PCR就可將模板DNA在體外大量擴增。
設計引物應遵循以下原則:
①引物長度: 15-30bp,常用爲20bp左右。
②引物擴增跨度: 以200-500bp爲宜,特定條件下可擴增長至10kb的片段。
③引物鹼基:G+C含量以40-60%爲宜,G+C太少擴增效果不佳,G+C過多易出現非特異條帶。ATGC最好隨機分佈,避免5個以上的嘌呤或嘧啶核苷酸的成串排列。
④避免引物內部出現二級結構,避免兩條引物間互補,特別是3’端的互補,否則會形成引物二聚體,產生非特異的擴增條帶。
⑤引物3’端的鹼基,特別是最末及倒數第二個鹼基,應嚴格要求配對,以避免因末端鹼基不配對而導致PCR失敗。
⑥引物中有或能加上合適的酶切位點,被擴增的靶序列最好有適宜的酶切位點,這對酶切分析或分子克隆很有好處。
⑦引物的特異性:引物應與核酸序列數據庫的其它序列無明顯同源性。
引物量: 每條引物的濃度0.1~1umol或10~100pmol,以最低引物量產生所需要的結果爲好,引物濃度偏高會引起錯配和非特異性擴增,且可增加引物之間形成二聚體的機會。
酶及其濃度 目前有兩種Taq DNA聚合酶供應, 一種是從棲熱水生桿菌中提純的天然酶,另一種爲大腸菌合成的基因工程酶。催化一典型的PCR反應約需酶量2.5U(指總反應體積爲100ul時),濃度過高可引起非特異性擴增,濃度過低則合成產物量減少。
dNTP的質量與濃度 dNTP的質量與濃度和PCR擴增效率有密切關係,dNTP粉呈顆粒狀,如保存不當易變性失去生物學活性。dNTP溶液呈酸性,使用時應配成高濃度後,以1M NaOH或1M Tris。HCL的緩衝液將其PH調節到7.0~7.5,小量分裝, -20℃冰凍保存。多次凍融會使dNTP降解。在PCR反應中,dNTP應爲50~200umol/L,尤其是注意4種dNTP的濃度要相等( 等摩爾配製),如其中任何一種濃度不同於其它幾種時(偏高或偏低),就會引起錯配。濃度過低又會降低PCR產物的產量。dNTP能與Mg2+結合,使遊離的Mg2+濃度降低。
模板(靶基因)核酸 模板核酸的量與純化程度,是PCR成敗與否的關鍵環節之一,傳統的DNA純化方法通常採用SDS和蛋白酶K來消化處理標本。SDS的主要功能是:溶解細胞膜上的脂類與蛋白質,因而溶解膜蛋白而破壞細胞膜,並解離細胞中的核蛋白,SDS 還能與蛋白質結合而沉澱;蛋白酶K能水解消化蛋白質,特別是與DNA結合的組蛋白,再用有機溶劑酚與氯仿抽提掉蛋白質和其它細胞組份,用乙醇或異丙醇沉澱核酸。提取的核酸即可作爲模板用於PCR反應。一般臨牀檢測標本,可採用快速簡便的方法溶解細胞,裂解病原體,消化除去染色體的蛋白質使靶基因遊離,直接用於PCR擴增。RNA模板提取一般採用異硫氰酸胍或蛋白酶K法,要防止RNase降解RNA。
Mg2+濃度 Mg2+對PCR擴增的特異性和產量有顯著的影響,在一般的PCR反應中,各種dNTP濃度爲200umol/L時,Mg2+濃度爲1.5~2.0mmol/L爲宜。Mg2+濃度過高,反應特異性降低,出現非特異擴增,濃度過低會降低Taq DNA聚合酶的活性,使反應產物減少。
PCR反應條件的選擇
PCR反應條件爲溫度、時間和循環次數。
溫度與時間的設置: 基於PCR原理三步驟而設置變性-退火-延伸三個溫度點。在標準反應中採用三溫度點法,雙鏈DNA在90~95℃變性,再迅速冷卻至40 ~60℃,引物退火並結合到靶序列上,然後快速升溫至70~75℃,在Taq DNA 聚合酶的作用下,使引物鏈沿模板延伸。對於較短靶基因(長度爲100~300bp時)可採用二溫度點法, 除變性溫度外、退火與延伸溫度可合二爲一,一般採用94℃變性,65℃左右退火與延伸(此溫度Taq DNA酶仍有較高的催化活性)。
①變性溫度與時間:變性溫度低,解鏈不完全是導致PCR失敗的最主要原因。一般情況下,93℃~94℃lmin足以使模板DNA變性,若低於93℃則需延長時間,但溫度不能過高,因爲高溫環境對酶的活性有影響。此步若不能使靶基因模板或PCR產物完全變性,就會導致PCR失敗。
②退火(復性)溫度與時間:退火溫度是影響PCR特異性的較重要因素。變性後溫度快速冷卻至40℃~60℃,可使引物和模板發生結合。由於模板DNA 比引物複雜得多,引物和模板之間的碰撞結合機會遠遠高於模板互補鏈之間的碰撞。退火溫度與時間,取決於引物的長度、鹼基組成及其濃度,還有靶基序列的長度。對於20個核苷酸,G+C含量約50%的引物,55℃爲選擇最適退火溫度的起點較爲理想。引物的復性溫度可通過以下公式幫助選擇合適的溫度:
Tm值(解鏈溫度)=4(G+C)+2(A+T)
復性溫度=Tm值-(5~10℃)
在Tm值允許範圍內, 選擇較高的復性溫度可大大減少引物和模板間的非特異性結合,提高PCR反應的特異性。復性時間一般爲30~60sec,足以使引物與模板之間完全結合。
③延伸溫度與時間:Taq DNA聚合酶的生物學活性:
70~80℃ 150核苷酸/S/酶分子
高於90℃時, DNA合成幾乎不能進行。
PCR反應的延伸溫度一般選擇在70~75℃之間,常用溫度爲72℃,過高的延伸溫度不利於引物和模板的結合。PCR延伸反應的時間,可根據待擴增片段的長度而定,一般1Kb以內的DNA片段,延伸時間1min是足夠 的。3~4kb的靶序列需3~4min;擴增10Kb需延伸至15min。延伸進間過長會導致非特異性擴增帶的出現。對低濃度模板的擴增,延伸時間要稍長些。
循環次數 循環次數決定PCR擴增程度。PCR循環次數主要取決於模板DNA的濃度。一般的循環次數選在30~40次之間,循環次數越多,非特異性產物的量亦隨之增多。
4.2 PCR反應條件的選擇及優化
PCR技術已經在生物學研究和臨牀醫學檢驗領域中得到了廣泛的應用,PCR技術也日臻完善。PCR技術操作簡便,特異性強,敏感度極高,正因爲敏感度高,很容易受其他因素的影響,因此要得到準確可靠的反應結果,需根據不同的模板,摸索最適合的條件,配製出PCR反應試劑。
聚合酶鏈反應必須具備下述基本條件:①模板核酸(DNA或RNA),②人工合成的寡核苷酸引物,③合適的緩衝體系,④鎂離子,⑤三磷酸脫氧核苷酸,⑥耐熱DNA聚合酶,RNA反轉錄酶及RNA酶抑制劑(RNasin),⑦溫度循環參數(變性、復性和延伸的溫度與時間及循環次數)。
1.模板的選擇
PCR反應用DNA(單、雙鏈DNA)或RNA都可以作模板進行核酸的體外擴增。若起始材料是RNA,須先通過逆轉錄得到cDNA後才能進行正常PCR擴增。核酸標本來源廣泛,可以從純培養的細胞或微生物中直接提取,也可以從臨牀標本(血、尿、便、痰、體腔積液、漱口水等)、犯罪現場標本(血斑、精斑、毛髮等)、病理標本(新鮮或固定石蠟包埋標本)以及木乃伊標本中提取。無論標本來源如何,待擴增核酸都需部分純化,以使核酸樣品中不混有任何蛋白酶、核酸酶、Taq DNA聚合酶抑制劑以及能結合DNA的蛋白質。雖然PCR可以僅用微量樣品(甚至是來自單一細胞的DNA),但爲保證反應的特異性,一般還宜用納克級(ng)的克隆DNA、微克水平的染色體DNA或102-105拷貝的待擴增DNA片段做起始材料。
2.引物
PCR擴增產物的大小及擴增的靶序列在基因組中的位置是由引物限定的。因此,引物的選擇與合成對PCR成功與否具有決定性意義。對某一DNA片段來說,由於同源順序的存在,隨意設計的兩條引物鏈,其PCR產物經電泳分析時可能會出現多條電泳帶。因此,在引物的設計過程中要考慮引物鏈的特異性,即它們只與被檢測的DNA片斷互補。
(1) 引物長度以16-30個鹼基爲宜,最佳20-24個bp,不會形成穩定的複合體。
(2) 有時引物不起作用,理由不明,可以移動序列位置來解決。
(3) (G+C)%含量一般40%-60%爲佳,兩個引物中(G+C)%含量應儘量相似。
(4) 兩個引物之間不應發生互補,特別是在引物3’端避免形成“引物二聚體”。如無法避免,其3’端互補不應大於2個鹼基。應儘量避免數個嘌呤或嘧啶的連續排列,避免同源序列(尤其6個以上相同)。
(5) 引物內部避免形成次級結構,尤其是髮卡結構,必要時應通過計算機進行結構分析。
(6) 引物濃度不宜偏高,濃度過高有兩個弊端,一是易形成引物二聚體,二是當擴增微量靶目標並且起始材料又不太純時容易產生非特異性產物。一般來說,用低濃度引物不僅經濟,反應特異性也較好。
3.鎂離子濃度
Mg2+濃度對PCR擴增反應的特異性和產量有着顯著影響。PCR中常用的體系應Mg2+濃度爲1.5mmol/L,但對不同的反應體系應優化Mg2+濃度。濃度過高,使反應特異性降低;濃度過低,使產物產量減少。PCR反應中Mg2+的加入量要比dNTP濃度高0.5-2.5mmol/L。最好對每種PCR反應體系Mg2+量的優化。另外,還需注意引物和模板DNA原液中是否含EDTA等螯合劑影響遊離Mg2+的濃度。
PCR反應中每種dNTP的終濃度爲50-200μmol/L,在此範圍內,擴增產物量、特異性與合成忠實性這間的平衡最佳,dNTP濃度過低必然影響擴增產量,過高則會導致錯誤摻入,其濃度不能低於10-15μmol/L。由於dNTP的量還受其他因素的影響,所以不同反應體系中dNTP的最佳濃度不盡相同。
5、TapDNA聚合酶
在其他參數最佳時,每100μl反應液中含1-2.5u Taq DNa酶。然而酶的需要量可以根據不同的模板分子或引物而變化,當優化一種PCR反應體系時,最好在每100μl體積中加入0.5-5u酶的範圍內試驗最佳酶濃度。如酶濃度太高,會出現非特異性擴增,而過低時,則擴增產量太低。另外不同來源的Taq DNA酶、測定條件和單位定義的不同、生產廠家產品品質的優劣,這些都是使用Taq酶時需要考慮的因素。
6.溫度循環參數
變性溫度通常爲94℃左右,主要取決於擴增片段的長度和(G+C)的比例,達不到變性溫度致使變性不完全,是導致PCR失敗的常見原因。變性時間一般爲30-60秒,時間太長不但沒有必要反而會因降低酶的活性而影響產量和反應特異性。常用PCR一般爲20-40個週期,隨着週期次數增加TaqDNA聚合酶活性降低,聚合時間延長,引物或單核苷酸減少等原因,會造成反應後期容易發生錯誤鹼基摻入。所以在滿足產物得率的前提下,應儘量減少週期次數。
7.石蠟油
加入石蠟油的目的是防止反應液蒸發,以免造成反應體積和成分的改變,特別是當反應體積小或反應條件嚴格時更應注意。反應中加入的石蠟油必須是高質量的,無菌的,不能含有抑制PCR反應中各種試劑活性的污染物,加入的體積要求不太嚴格,應以蓋住反應液液麪爲度。
8.平臺效應
“平臺效應”是指PCR後期循環產物累積趨於飽和,使原來以指數增長的速率變成平坦的曲線,同時出現非特異產物的大量增加的現象。下列因素可能與平臺效應有關:
(1)dNTP或引物的消耗量。
(4)非特異產物或引物二聚體與反應物的競爭。
(5)產物在高濃度時變性不完全,影響引物的延伸。
(6)酶與PCR產物的結合,使酶分子減少。
合理的PCR循環參數,能最大限度地避免這些因素對產物擴增的影響。
4.3 PCR反應的常見問題總結
PCR 的問題,無疑是絕大多數學生物的人都關心的問題。今天我們把相關內容整理出來,與大家共同討論,希望對大家能有所幫助。
一、引物設計
所謂“工欲善其事,必先利其器”,這年頭手工設計引物的人似乎不多,還是用軟件方便些,防止你一不小心看走眼,丟一個鹼基,同時計算起來也方便。設計軟件有很多,既可以在線設計(如Primer3 http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi),也可以用Primer5、Oligo6.65 等等。
1. 引物設計的原則 細心地進行引物設計是PCR 中最重要的一步。理想的引物對只同目的序列兩側的單一序列而非其他序列退火。設計糟糕的引物可能會同時擴增其他的非目的序列。下面的指導描述了一個可以增加特異性的引物所具有的令人滿意的特點: 1)典型的引物18 到24 個核苷長。引物需要足夠長,保證序列獨特性,並降低序列存在於非目的序列位點的可能性。但是長度大於24 核苷的引物並不意味着更高的特異性。較長的序列可能會與錯誤配對序列雜交,降低了特異性,而且比短序列雜交慢,從而降低了產量。 2)選擇GC 含量爲40%到60%或GC 含量與模板GC 含量接近的引物。 3)設計5'端和中間區爲G 或C 的引物。這會增加引物的穩定性和引物同目的序列雜交的穩定性。 4)避免引物對3'末端存在互補序列,這會形成引物二聚體,抑制擴增。在用軟件設計時大家常常會疑惑,究竟? G 不能低於多少?這裏有個數據: ? G 爲0~-2時,PCR 產率可達100%, ? G=-6 時,爲40%. 5)避免3'末端富含GC。設計引物時保證在最後5 個核苷中含有3 個A 或T。 6)避免3'末端的錯誤配對。3'端核苷需要同模板退火以供聚合酶催化延伸。3’最好以G或C 結尾,防止AT 的鬆散結合引起錯配。 7)避免存在可能會產生內部二級結構如髮夾結構的序列,這會破壞引物退火穩定性。 8)引物的一個重要參數是熔解溫度(Tm)。這是當50%的引物和互補序列表現爲雙鏈DNA 分子時的溫度。兩引物的Tm 值相差不應大於5℃。計算Tm 有幾種公式。第一個公式(Wallace 規則):Tm = 4℃(g + C) +2℃(a + T),這來源於高鹽溶液中的雜交,適用於小於15-20個鹼基的引物,也適用於手工設計時的簡單計算。第二個公式(Baldino 算法)適用於計算14-70個核苷酸在≤0.4mol\L 的陽離子溶液中的Tm, 也可用於擴增產物的Tm 計算.Tm=81.5+16.6*lg[K+]+0.41(%[G+C])-(675/n)。以上兩種算法都是基於鹼基組成而不是鹼基排列而計算的,事實上相同鹼基組成的引物Tm 可能差異不小:GGGAA 和GAGAG 的Tm 是不一樣的,所以確定引物Tm 最可信的方法是近鄰分析法。這種方法從序列一級結構和相鄰鹼基的特性預測引物的雜交穩定性。大部分計算機程序如Primer5 等均使用近鄰分析法。 9)當在引物5’端添加酶切位點時要考慮:a)該目的序列內部不得含有相同的酶切位點,在引物發出後才發現錯誤的事情本人就幹過,在論壇上也能看到這樣的粗心人。這樣的錯誤會給將來的克隆造成麻煩。b)如果打算PCR 後直接酶切,不要忘了在酶切位點的外側再加上保護鹼基,不同的酶對於保護鹼基的要求是不同的。如果不設計保護鹼基,則多半要用TA 克隆的方式連接到質粒上,這時要注意Taq 酶的選擇,這一點在後面再聊。若想在目的序列上附加上並不存在的序列,如限制位點和啓動子序列,可以加入到引物5'端而不影響特異性。當計算引物Tm 值時並不包括這些序列,但是應該對其進行互補性和內部二級結構的檢測。 10)有時候,對於引物設計僅瞭解有限的序列信息。比如,如果僅知道氨基酸序列,可以設計兼併引物。兼併引物是指代表編碼單個氨基酸所有不同鹼基可能性的不同序列的混合物。爲了增加特異性,可以參考密碼子使用表,根據不同生物的鹼基使用偏好,減少兼併性。次黃嘌呤可以同所有的鹼基配對,降低引物的退火溫度。特別要注意的是:不要在引物的3'端使用兼併鹼基,因爲3'端最後3 個鹼基的退火足以在錯誤位點起始PCR。使用較高的引物濃度(1µM 到3µM),因爲許多兼併混合物中的引物不是特異性針對目的模板。說了半天了,想起來還得提醒大家一句:千萬別搞錯了引物的位置!這句話似乎多餘。仔細琢磨一下,特別是5’端再接上酶切位點可別搞錯了!合成錯了一條可就是50 塊錢,到時候挨老闆批的時候別怪我沒提醒你哦:)設計好了引物就要讓公司合成,這時候別忘了談價格:)現在競爭很厲害,價格已經挺便宜了,注意兩個參數:一個是OD 值,如果公司說1OD 能比2OD 優惠,那通常1OD 足夠了。第二是純度,是HPLC 的還是OPC 的,事先要說好,建議看看這裏(唉,不是我給他們做廣告,只是他們的介紹寫得真不錯):http://www.takara.com.cn/product/cs/c_3.htm#1引物設計方面還有很多話題可說,比如如何利用引物搭橋將兩個片段通過PCR 而不是酶連接起來、如果要設計引物表達蛋白時注意“N 端規則”。
二、PCR 成分
引用《分子克隆3》提供的PCR 標準反應條件: 模板1pg-1μg 引物1μmol/L DNA 聚合酶1-5 單位 Mg2+1.5mmol/L dNTPs 200μmol/L KCl 50 mmol/L
1. 模板 模板可是PCR 的關鍵,模板的質量是PCR 成功的先決條件,巧婦難爲無米之炊嘛!怎樣得到模板,這可是一個大問題,僅僅一個提取基因組DNA 就有很多的名堂,這會是我們後面專輯的內容,這裏不多說啦,有問題來論壇討論吧。P 不出東西的時候不妨先考慮:模板有沒有問題?(DNA 樣品中發現有多種污染物會抑制PCR。一些在標準基因組DNA 製備中使用的試劑,如SDS,在濃度低至0.01%時就會抑制擴增反應。)所需的最佳模板量取決於基因組的大小。你的目的片段在基因組中佔多少?至少要保證模板中含有一個單拷貝吧!100ng 到1µg 的人類基因組DNA,相當於3×104 到3×105 個分子。所以對於單拷貝基因,這需要0.1μg 的人基因組DNA,10ng的酵母DNA,1ng 的大腸桿菌DNA.擴增多拷貝序列時,用量更少.靈敏的PCR 可從一個細胞,一根頭髮,一個孢子或一個精子提取的DNA中分析目的序列.模板量過多則可能增加非特異性產物.DNA中的雜質也會影響PCR的效率。
2. 引物 引物以乾粉形式運輸。最好用TE 重溶引物,使其最終濃度爲100µM。TE 比去離子水好,因爲水的pH 經常偏酸,會引起寡核苷的水解。當然,如果擔心TE 對於PCR 的影響,不妨用ddH2O。引物的穩定性依賴於儲存條件。應將乾粉和溶解的引物儲存在-20℃。以大於10µM 濃度溶於TE 的引物在-20℃可以穩定保存6 個月,但在室溫(15℃到30℃)僅能保存不到1 周。乾粉引物可以在-20℃保存至少1 年,在室溫(15℃到30℃)最多可以保存2 個月。注意:溶解之前先離心一下,防止一開蓋就飛了。引物的濃度會影響特異性。最佳的引物濃度一般在0.1 到0.5µM。較高的引物濃度會導致非特異性產物擴增。通常跟着引物來的介紹會告訴你,關於寡核苷酸的計算可以看看這裏:http://www.takara.com.cn/product/fl/i_1.htm
3. 聚合酶的選擇 這可是有講究,咱論壇裏有個專題討論這個問題。主要考慮兩個方面:用途——對保真度的要求高不高?成本——高保真的酶總是會貴一些的。綜合考慮一下找個平衡點吧,當然啦,該花錢的時候可不能省。Taq DNA聚合酶被看做是低保真度的聚合酶,因爲其缺少3'到5'外切核酸酶(校正)活性。使用帶有3'到5'外切核酸酶活性的熱穩定聚合酶可以提高保真度。但是這些聚合酶的產量比Taq DNA聚合酶低。在很多公司的手冊中有對各種酶特性的描述,大家不妨比較一下。提醒一點:高保真酶除了我所知的Merck的Easy-A 以外,都不會在3’端加A尾巴(因爲其3’-5’外切酶活性),所以做TA克隆的時候需要一點小訣竅——擴增完了再加普通Taq去加個A。另外還有個方法:將Taq DNA聚合酶同帶有3'到5'外切核酸酶活性第二種聚合酶混合在一起可以獲得比單獨Taq DNA聚合酶高的保真度,並可以得到高產量及擴增長模板。 4. Mg2+濃度 鎂離子影響PCR的多個方面,如DNA聚合酶的活性,這會影響產量;再如引物退火,這會影響特異性。dNTP和模板同鎂離子結合,降低了酶活性所需要的遊離鎂離子的量。最佳的鎂離子濃度對於不同的引物對和模板都不同,但是包含200µM dNTP的典型PCR起始濃度是1.5mM(注意:對實時定量PCR,使用3到5mM帶有熒光探針的鎂離子溶液)。在多數情況下,較高的遊離鎂離子濃度可以增加產量,但也會增加非特異性擴增,降低忠實性(當然,也有相反的報道)。爲了確定最佳濃度, 可以用0.1-5mmol/L的遞增濃度的Mg2+ 進行預備實驗,選出最適的Mg2+濃度。在PCR反應混合物中, 應儘量減少有高濃度的帶負電荷的基團, 例如磷酸基團或EDTA等可能影響Mg2+ 離子濃度的物質,以保證最適Mg2+ 濃度。 5. dNTPs 高濃度的dNTPs會對擴增反應起抑制作用。將每種dNTP的濃度從200µM降低到25-50µM可以使擴增產物獲得滿意的產率。 6. KCl 標準濃度爲50 mmol/L, 對於較短片段可將其提高到70-100 mmol/L. 三、PCR 反應參數 1. 變性:在第一輪循環前,在94℃下變性5-10min 非常重要,它可使模板DNA 完全解鏈,然後加入Taq DNA聚合酶(hot start),這樣可減少聚合酶在低溫下仍有活性從而延伸非特異性配對的引物與模板複合物所造成的錯誤。變性不完全,往往使PCR 失敗,因爲未變性完全的DNA 雙鏈會很快復性,減少DNA 產量.一般變性溫度與時間爲94℃ 1min。在變性溫度下,雙鏈DNA 解鏈只需幾秒鐘即可完全,所耗時間主要是爲使反應體系完全達到適當的溫度。對於富含GC 的序列,可適當提高變性溫度.但變性溫度過高或時間過長都會導致酶活性的損失。 2. 退火:這是PCR 的一個關鍵參數。在理想狀態下,退火溫度足夠低,以保證引物同目的序列有效退火,同時還要足夠高,以減少非特異性結合。合理的退火溫度從55℃到70℃。退火溫度一般設定比引物的Tm 低5℃, 當產物中包含有影響實驗的非可異性擴增帶時,以2℃爲增量,逐步提高退火溫度。較高的退火溫度會減少引物二聚體和非特異性產物的形成。如果兩個引物Tm 不同,將退火溫度設定爲比最低的Tm 低5℃。或者爲了提高特異性,可以在根據較高Tm 設計的退火溫度先進行5 個循環,然後在根據較低Tm 設計的退火溫度進行剩餘的循環。這使得在較爲嚴緊的條件下可以獲得目的模板的部分拷貝。退火溫度越高,所得產物的特異性越高。有些反應甚至可將退火與延伸兩步合併,只用兩種溫度(例如用60℃和94℃)完成整個擴增循環, 既省時間又提高了特異性。退火一般僅需數秒鐘即可完成,反應中所需時間主要是爲使整個反應體系達到合適的溫度。 3. 延伸:延伸反應通常爲72℃,接近於Taq DNA 聚合酶的最適反應溫度75℃.實際上,引物延伸在退火時即已開始,因爲Taq DNA 聚合酶的作用溫度範圍可從20℃-85℃.延伸反應時間的長短取決於目的序列的長度和濃度.在一般反應體系中,Taq DNA聚合酶每分鐘約可合成1kb 長的DNA。延伸時間過長會導致產物非特異性增加.但對很低濃度的目的序列, 則可適當增加延伸反應的時間。一般在擴增反應完成後,都需要一步較長時間(10-30min)的延伸反應,以獲得儘可能完整的產物, 這對以後進行克隆或測序反應尤爲重要。
4. 循環次數: 當其它參數確定之後, 循環次數主要取決於DNA 濃度。一般而言25-30輪循環已經足夠。循環次數過多,會使PCR 產物中非特異性產物大量增加。通常經25-30 輪循環擴增後, 反應中Taq DNA 聚合酶已經不足, 如果此時產物量仍不夠, 需要進一步擴增,可將擴增的DNA 樣品稀釋103-105倍作爲模板, 重新加入各種反應底物進行擴增, 這樣經60 輪循環後, 擴增水平可達109-1010。擴增產物的量還與擴增效率有關,擴增產物的量可用下列公式表示:C=Co(1+P)n。其中:C 爲擴增產物量,C0 爲起始DNA 量, P 爲增效率, n 爲循環次數。在擴增後期,由於產物積累,使原來呈指數擴增的反應變成平坦的曲線,產物不再隨循環數而明顯上升,這稱爲平臺效應。平臺期會使原先由於錯配而產生的低濃度非特異性產物繼續大量擴增,達到較高水平。因此,應適當調節循環次數,在平臺期前結束反應,減少非特異性產物。
四、提高PCR 擴增特異性
1. 遞減PCR(TouchDown PCR) 遞減PCR 通過在PCR 的前幾個循環使用嚴緊的退火條件提高特異性。循環設在比估算的Tm 高大約5℃的退火溫度下開始,然後每個循環降低1℃到2℃(當然,也可以每幾個循環降1-2℃),直到退火溫度低於Tm 5℃。特異性最高的目的模板會被優先擴增,這些產物在隨後的循環中繼續擴增佔據優勢。遞減PCR 對於那些不瞭解引物和目的模板同源性程度的方法更爲有用,如AFLP® DNA 指紋分析。
2. 熱啓動 熱啓動PCR 是除了好的引物設計之外,提高PCR 特異性最重要的方法之一。儘管TaqDNA 聚合酶的最佳延伸溫度在72℃,聚合酶在室溫仍然有活性。因此,在進行PCR 反應配製過程中,以及在熱循環剛開始,保溫溫度低於退火溫度時會產生非特異性的產物。這些非特異性產物一旦形成,就會被有效擴增。在用於引物設計的位點因爲遺傳元件的定位而受限時,如定點突變、表達克隆或用於DNA 工程的遺傳元件的構建和操作,熱啓動PCR 尤爲有效。並且,熱啓動在很大程度上防止引物二聚的發生。限制Taq DNA 聚合酶活性的常用方法是在冰上配製PCR 反應液,並將其置於預熱的PCR 儀。這種方法簡單便宜,但並不能完成抑制酶的活性,因此並不能完全消除非特異性產物的擴增。熱啓動通過抑制一種基本成分延遲DNA 合成,直到PCR 儀達到變性溫度。包括延緩加入Taq DNA 聚合酶在內的大部分手工熱啓動方法十分煩瑣,尤其是對高通量應用。其他的熱啓動方法使用蠟防護層將一種基本成分,入鎂離子或酶,包裹起來,或者將反應成分,如模板和緩衝液,物理地隔離開。在熱循環時,因蠟熔化而把各種成分釋放出來並混合在一起。象手動熱啓動方法一樣,蠟防護層法比較煩瑣,易於污染,不適用于于高通量應用。
3. 促進PCR 的添加劑 退火溫度,引物設計和鎂離子濃度的優化足以對大多數模板進行高特異性的擴增,但是,某些模板,包括高GC 含量的模板,需要其他的措施。影響DNA 熔解溫度的添加劑提供了提高產物特異性和產量的另外一種方法。爲獲得最好的結果需要模板的完全變性。另外,二級結構會阻止引物結合和酶的延伸。PCR 添加劑,包括甲酰胺,DMSO,甘油,甜菜鹼以及PCRx Enhancer Solution 可以增強擴增。它們可能的機理是降低熔解溫度,從而有助於引物退火並輔助DNA 聚合酶延伸通過二級結構區。PCRx Solution 還有其他優點。在同PlatinumTaq DNA 聚合酶和Platinum Pfx DNA 聚合酶一起使用時,僅需很少的鎂離子優化。這樣,將Platinum 技術同添加劑結合,增強了特異性,同時減少了第三種方法-鎂離子優化的依賴。爲獲得最佳結果,應優化添加劑的濃度,尤其是會抑制Taq DNA 聚合酶的DMSO,甲酰胺和甘油。
4. 巢式PCR 使用巢式引物進行連續多輪擴增可以提高特異性和靈敏度。第一輪是15 到20 個循環的標準擴增。將一小部分起始擴增產物稀釋100 到1000 倍加入到第二輪擴增中進行15 到20個循環。或者,也可以通過凝膠純化將起始擴增產物進行大小選擇。在第二輪擴增中使用一套巢式引物,其可以同第一套引物內側的靶序列結合。巢式PCR 的使用降低了擴增多個靶位點的可能性,因爲同兩套引物都互補的靶序列很少。而使用同樣的引物對進行總數相同的循環(30 到40)會擴增非特異性靶位點。巢式PCR 可以增加有限量靶序列(如稀有mRNA)的靈敏度,並且提高了困難PCR(如5' RACE)的特異性。
五、PCR 產物測序
對於PCR 產物的測序有兩個方式:
1. DNA 直接測序: 指直接分析不經分離的PCR 擴增產物,如果各個位點上鹼基序列都是一致的,則所得信號是確定的,否則,在那些有相異鹼基的位點出現模糊信號, 如果模板在多數情況下被正確擴增,則少數的突變將被掩蓋,這是結果顯示的是模板的序列.但是,當擴增的前幾輪即出現錯誤擴增並被後續的循環積累,這時就不再反映模板的狀況.
2. PCR 產物經克隆後測序: 這是對單克隆測序,其結果是反映單一擴增產物的序列,結果是確定的.
六、PCR 污染與對策
PCR 反應的最大特點是具有較大擴增能力與極高的靈敏性,但令人頭痛的問題是易污染,極其微量的污染即可造成假陽性的產生.
1、污染原因 (一)標本間交叉污染:標本污染主要有收集標本的容器被污染,或標本放置時,由於密封不嚴溢於容器外,或容器外粘有標本而造成相互間交叉污染;標本核酸模板在提取過程中,由於吸樣槍污染導致標本間污染;有些微生物標本尤其是病毒可隨氣溶膠或形成氣溶膠而擴散,導致彼此間的污染. (二)PCR 試劑的污染:主要是由於在PCR 試劑配製過程中,由於加樣槍、容器、雙蒸水及其它溶液被PCR 核酸模板污染. (三)PCR 擴增產物污染.這是PCR 反應中最主要最常見的污染問題.因爲PCR 產物拷貝量大(一般爲1013 拷貝/ml),遠遠高於PCR 檢測數個拷貝的極限,所以極微量的PCR 產物污染,就可造成假陽就可形成假陽性. 還有一種容易忽視,最可能造成PCR 產物污染的形式是氣溶膠污染;在空氣與液體面摩擦時就可形成氣溶膠,在操作時比較劇烈地搖動反應管,開蓋時、吸樣時及污染進樣槍的反復吸樣都可形成氣溶膠而污染.據計算一個氣溶膠顆粒可含48000 拷貝,因而由其造成的污染是一個值得特別重視的問題. (四)實驗室中克隆質粒的污染:在分子生物學實驗室及某些用克隆質粒做陽性對照的檢驗室,這個問題也比較常見.因爲克隆質粒在單位容積內含量相當高,另外在純化過程中需用較多的用具及試劑,而且在活細胞內的質粒,由於活細胞的生長繁殖的簡便性及具有很強的生命力.其污染可能性也很大.
2、污染的監測 一個好的實驗室,要時刻注意污染的監測,考慮有無污染是什麼原因造成的污染,以便採取措施,防止和消除污染. 對照試驗: 1)陽性對照:在建立PCR 反應實驗室及一般的檢驗單位都應設有PCR 陽性對照,它是PCR 反應是否成功、產物條帶位置及大小是否合乎理論要求的一個重要的參考標誌.陽性對照要選擇擴增度中等、重複性好,經各種鑑定是該產物的標本,如以重組質粒爲陽性對照,其含量宜低不宜高(100 個拷貝以下).但陽性對照尤其是重組質粒及高濃度陽性標本,其對檢測或擴增樣品污染的可能性很大.因而當某一PCR 試劑經自己使用穩定,檢驗人員心中有數時,在以後的實驗中可免設陽性對照. 2)陰性對照:每次PCR 實驗務必做陰性對照.它包括①標本對照:被檢的標本是血清就用鑑定後的正常血清作對照;被檢的標本是組織細胞就用相應的組織細胞作對照.②試劑對照:在PCR 試劑中不加模板DNA 或RNA,進行PCR 擴增,以監測試劑是否污染. 3)重複性試驗 4)選擇不同區域的引物進行PCR 擴增
3、防止污染的方法 (一)合理分隔實驗室:將樣品的處理、配製PCR 反應液、PCR 循環擴增及PCR 產物的鑑定等步驟分區或分室進行,特別注意樣本處理及PCR 產物的鑑定應與其它步驟嚴格分開.最好能劃分①標本處理區;②PCR 反應液製備區;③PCR 循環擴增區;④PCR 產物鑑定區. 其實驗用品及吸樣槍應專用.實驗前應將實驗室用紫外線消毒以破壞殘留的DNA 或RNA. (二)吸樣槍:吸樣槍污染是一個值得注意的問題.由於操作時不慎將樣品或模板核酸吸入槍內或粘上槍頭是一個嚴重的污染源,因而加樣或吸取模板核酸時要十分小心,吸樣要慢,吸樣時儘量一次性完成,忌多次抽吸,以免交叉污染或產生氣溶膠污染. (三)預混和分裝PCR 試劑:所有的PCR 試劑都應小量分裝,如有可能,PCR 反應液應預先配製好,然後小量分裝,-20℃保存.以減少重複加樣次數,避免污染機會.另外,PCR 試劑,PCR 反應液應與樣品及PCR 產物分開保存,不應放於同一冰盒或同一冰箱. (四)防止操作人員污染,使用一次性手套、吸頭、小離心管應一次性使用. (五)設立適當的陽性對照和陰性對照,陽性對照以能出現擴增條帶的最低量的標準病原體核酸爲宜,並注意交叉污染的可能性,每次反應都應有一管不加模板的試劑對照及相應不含有被擴增核酸的樣品作陰性對照. (六)減少PCR 循環次數,只要PCR 產物達到檢測水平就適可而止. (七)選擇質量好的Eppendorf 管,以避免樣本外溢及外來核酸的進入,打開離心管前應先離心,將管壁及管蓋上的液體甩至管底部.開管動作要輕,以防管內液體濺出.
4、PCR 污染解決對策(這是從另一篇文章總結而來,與前面的部分略有重複,大家隨便看看吧)PCR 檢測微量感染因子時,一定要注意產物殘留污染的問題。 一. 污染的預防 進行PCR 操作時,操作人員應該嚴格遵守一些操作規程,最大程度地降低可能出現的 PCR 污染或杜絕污染的出現。 (一)劃分操作區:目前,普通PCR 尚不能做到單人單管,實現完全閉管操作,但無論是否能夠達到單人單管,均要求實驗操作在三個不同的區域內進行,PCR 的前處理和後處理要在不同的隔離區內進行: 1. 標本處理區,包括擴增摸板的製備; 2. PCR 擴增區,包括反應液的配製和PCR 擴增; 3. 產物分析區,凝膠電泳分析,產物拍照及重組克隆的製備。 各工作區要有一定的隔離,操作器材專用,要有一定的方向性。如:標本製備→PCR擴增? 產物分析? 產物處理。 切記:產物分析區的產物及器材不要拿到其他兩個工作區。 (二)分裝試劑:PCR 擴增所需要的試劑均應在裝有紫外燈的超淨工作臺或負壓工作臺配製和分裝。所有的加樣器和吸頭需固定放於其中,不能用來吸取擴增後的DNA 和其他來源 的DNA: 1.PCR 用水應爲高壓的雙蒸水; 2.引物和dNTP 用高壓的雙蒸水在無PCR 擴增產物區配製; 3.引物和dNTP 應分裝儲存,分裝時應標明時間,以備發生污染時查找原因。 (三) 實驗操作注意事項儘管擴增序列的殘留污染大部分是假陽性反應的原因,樣品間的交叉污染也是原因之一。因此,不僅要在進行擴增反應是謹慎認真,在樣品的收集、抽提和擴增的所有環節都應該注意: 1. 戴一次性手套,若不小心濺上反應液,立即更換手套; 2. 使用一次性吸頭,嚴禁與PCR 產物分析室的吸頭混用,吸頭不要長時間暴露於空氣中,避免氣溶膠的污染; 3. 避免反應液飛濺,打開反應管時爲避免此種情況,開蓋前稍離心收集液體於管底。若不小心濺到手套或桌面上,應立刻更換手套並用稀酸擦拭桌面; 4. 操作多份樣品時,製備反應混合液,先將dNTP、緩衝液、引物和酶混合好,然後分裝,這樣即可以減少操作,避免污染,又可以增加反應的精確度; 5. 最後加入反應模板,加入後蓋緊反應管; 6. 操作時設立陰陽性對照和空白對照,即可驗證PCR 反應的可靠性,又可以協助判斷擴增系統的可信性; 7. 儘可能用可替換或可高壓處理的加樣器,由於加樣器最容易受產物氣溶膠或標本DNA 的污染,最好使用可替換或高壓處理的加樣器。如沒有這種特殊的加樣器,至少PCR操作過程中加樣器應該專用,不能交叉使用,尤其是PCR 產物分析所用加樣器不能拿到其它兩個區; 8. 重複實驗,驗證結果,慎下結論。 二. 追蹤污染源 如果不慎發生污染情況,應從下面幾條出發,逐一分析,排除污染。 (一)設立陰陽性對照:有利於監測反應體系各成分的污染情況。選擇陽性對照時,應選擇擴增弱,且重複性好的樣品,因強陽性對照可產生大量不必要的擴增序列,反而可能成爲潛在的污染源。如果以含靶序列的重組質粒爲對照,100 個拷貝之內的靶序列就足以產生陽性擴增。陰性對照的選擇亦要慎重,因爲PCR 敏感性極高,可以從其它方法(Sourthern 印跡或點雜交等)檢測陰性的標本中檢測出極微量的靶分子。此外,每次擴增均應包括PCR 體系中各試劑的時機對照,即包括PCR 反應所需的全部成分,而不加模板DNA,這對監測試劑中PCR 產物殘留污染是非常有益的。如果擴增結果中試劑對照爲陽性結果,就是某一種或數種試劑被污染了。此時,要全部更換一批新的試劑進行擴增,擴增時設立不同的反應管,每一管含有一種被檢測試劑,在檢出污染試劑後,應馬上處理。 (二)環境污染:在排除試劑污染的可能性外,更換試劑後,若不久又發現試劑被污染了,如果預防措施比較嚴密,則考慮可能爲環境污染。 環境污染中常見的污染源主要有: 1. 模板提取時真空抽乾裝置; 2. 凝膠電泳加樣器; 3. 電泳裝置; 4. 紫外分析儀; 5. 切膠用刀或手術刀片; 6. 離心機; 7. 冰箱門把手,冷凍架,門把手或實驗檯面等;此時可用擦拭實驗來查找可疑污染源。1)用無菌水浸泡過的滅菌棉籤擦拭可疑污染源;2)0.1ml 去離子水浸泡;3)取5ml 做PCR 實驗;4)電泳檢測結果。 8. 氣溶膠。如果經過上述追蹤實驗,仍不能查找到確切污染源,則污染可能是由空氣中PCR 產物的氣溶膠造成的,此時就應該更換實驗場所,若條件不允許,則重新設計新的引物(與原引物無相關性)。 三.污染處理 (一)環境污染 1. 稀酸處理法:對可疑器具用1mol/L 鹽酸擦拭或浸泡,使殘餘DNA 脫嘌呤; 2. 紫外照射(UV)法:紫外波長(nm)一般選擇254/300nm,照射30min 即可。需要注意的是,選擇UV 作爲消除殘留PCR 產物污染時,要考慮PCR 產物的長度與產物序列中鹼基的分佈,UV 照射僅對500bp 以上長片段有效,對短片段效果不大。UV 照射時,PCR產物中嘧啶鹼基會形成二聚體,這些二聚體可使延伸終止,但並不是DNA 鏈中所有嘧啶均能形成二聚體,且UV 照射還可使二聚體斷裂。形成二聚體的程度取決於UV 波長,嘧啶二聚體的類型及與二聚體位點相鄰核苷酸的序列。在受照射的長DNA 鏈上,形成二聚體缺陷的數量少於0.065/鹼基,其他非二聚體的光照損傷(如環丁烷型嘧啶複合體,胸腺嘧啶乙二醇,DNA 鏈間與鏈內的交聯和DNA 斷裂等)均可終止Taq DNA 聚合酶的延伸。這些位點的數量與二聚體位點相當。如果這些位點( 0.13/鹼基)在DNA 分子上隨機分佈,一個500bp片段的DNA 分子鏈上將有32 處損傷位點,那麼,105個這樣的分子中每個分子中會至少有一處損傷。相反,如果100bp 的片段,每條鏈上僅有6 處損傷,105個拷貝分子中將有許多分子沒有任何損傷。這就是UV 照射有一定的片段長度限制的原因。 (二)反應液污染 可採用下列方法之一處理: 1. DNase I 法:PCR 混合液(未加模板和Taq 聚合酶)加入0.5U DNase I,室溫反應30min 後加熱滅活,然後加入模板和Taq 聚合酶進行正常PCR 擴增。該方法的優點是不需要知道污染DNA 的序列; 2. 內切酶法:選擇識別4 個鹼基的內切酶(如Msp I 和Taq I 等),可同時選擇幾種,以克服用一種酶只能識別特定序列的缺陷,室溫作用1h 後加熱滅活進行PCR; 3. 紫外照射法:未加模板和Taq 聚合酶的PCR 混合液進行紫外照射,注意事項與方法同上述UV 照射法; 4. g 射線輻射法:1.5kGy 的輻射可完全破壞0.1ng 基因組DNA,2.0 kGy 可破壞104 拷貝的質粒分子,4.0 kGy 仍不影響PCR,但高於此限度會使PCR 擴增效率下降。引物可受照射而不影響PCR,g 射線是通過水的離子化產生自由基來破壞DNA 的。 (三)尿嘧啶糖苷酶(UNG)法 由於UV 照射的去污染作用對500bp 以下的片段效果不好,而臨牀用於檢測的PCR 擴增片段通常爲300bp 左右,因此UNG 的預防作用日益受到重視和肯定。 1. 原理:在PCR 產物或引物中用dU 代替dT。這種dU 化的PCR 產物與UNG 一起孵育,因UDG 可裂解尿嘧啶鹼基和糖磷酸骨架間的N-糖基鍵,可除去dU 而阻止TaqDNA 聚合酶的延伸,從而失去被再擴增的能力。UNG 對不含dU 的模板無任何影響。UNG 可從單或雙鏈DNA 中消除尿嘧啶,而對RNA 中的尿嘧啶和單一尿嘧啶分子則無任何作用。 2. dUTP 法:用dUTP 代替dTTP,使產物中摻入大量dU。在再次進行PCR 擴增前,用UNG 處理PCR 混合液即可消除PCR 產物的殘留污染。由於UNG 在PCR 循環中的變性一步便可被滅活,因此不會影響含dU 的新的PCR 產物。 3. dU 引物法:合成引物時以dU 代dT,這樣PCR 產物中僅5ˊ端帶dU。UNG 處理後,引物失去了結合位點而不能擴增。對長片段(1-2kb 以上)的擴增用dUTP 法效率較用dTTP低,而用dU 法就可克服這一缺點。dU 引物最好將dU 設計在3ˊ端或近ˊ端。該法僅能用於引物以外試劑的處理。 4. 優點:可以去除任何來源的污染;UNG 處理可以和PCR 擴增在同一個反應管內進行;由於擴增產物中有大量dU 存在,可徹底消除污染源。 5. 需注意的是摻入dUTP 的DNA不應對產物的任何操作有影響,在進行PCR 產物克隆時,應該轉化UNG-(UNG 缺陷)大腸桿菌受體菌,否則轉化產物會被受體菌UNG 消化掉。 (四) 固相捕獲法 用於去除標本中污染的核酸和雜質,原理如下:1)用一生物素標記的單鏈RNA 探針與待擴核酸雜交,雜交區域是非擴增區;2)用包被鏈黴親和素的固相載體來捕獲帶有生物素探針的雜交核酸,通過漂洗可去除污染的擴增產物和雜質;3)洗脫靶分子後用特異引物擴增非RNA 探針雜交區域。第2)步的漂洗後可用PCR 檢測以確定標本是否被擴增產物或重組質粒污染。 (五)RS-PCR 法(RNA-specific PCR) 也稱爲鏈特異性PCR,主要指用於RNA 模板的特異性PCR 法,該法可明顯降低假陽性而不影響PCR 的敏感性。其關鍵在於設計引物,逆轉錄引物的3ˊ端(A 區)有2 0 個核苷酸左右爲模板的特異性互不序列,5ˊ端2 0 個核苷酸(C 區)爲附加修飾鹼基。與mRNA逆轉錄後,經超速離心使cDNA 與多餘引物分開,再用和第二引物(C)以第一鏈cDNA爲模板合成第二鏈cDNA,以後的PCR 循環中用逆轉錄引物的B 區和引物C 進行擴增加尾cDNA,而污染的DNA 或質粒DNA 纔不會被擴增。 (六)抗污染引物法 該對引物擴增時通過病毒DNA克隆如入質粒的位點。這一區域只存在完整的原病毒中,在重組質粒中,這一區域分成兩個區域與克隆位點被。如果重組質粒污染了標本,也不能擴增出任何條帶,即使出現了擴增帶,其大小也與預期的不同。只有原病毒DNA 才能被引物擴增,因此只要出現預期大小的擴增帶就可以證明標本是陽性的,該法試用於環狀靶分子系列。
附錄: 各種鹼基符號
在設計簡併引物或測序時你可能會碰到這樣的符號來表示鹼基,到時候不要不知道哦: B = C or G or T D = A or G or T H = A or C or T K = G or T M = A or C N = A or C or G or T R = A or G S = C or G V = A or C or G W = A or T Y = C or T
5 結果檢測與分析
5.1 PCR擴增產物的檢測分析
PCR擴增反應完成之後,必須通過嚴格的鑑定,才能確定是否真正得到了準確可靠的預期特定擴增產物。凝膠電泳是檢測PCR產物常用和最簡便的方法,能判斷產物的大小,有助於產物的鑑定。凝膠電泳常用的有瓊脂糖凝膠電泳和聚丙烯酰胺凝膠電泳,前者主要用於DNA片段大於100bp者,後者主要用來檢測小片段DNA。
這是實驗室最常用的方法,簡便易行,只需少量DNA即可進行實驗。其原理是不同大小的DNA分子通過瓊脂糖凝膠時,由於泳動速度不同而被分離,經溴化乙錠(EB)染色,在紫外光照射下DNA分子發出熒光而判定其分子的大小。
用於電泳檢測PCR產物的瓊脂糖濃度常爲1%—2%,應該使用純度高的電泳純級瓊脂糖,這種瓊脂糖已除去了熒光抑制劑及核酸酶等雜質。
(1)制膠
瓊脂糖凝膠厚度約爲3~5mm。過薄則加樣孔樣品會溢出來,過厚觀察時熒光穿透不強以至有些小片段DNA帶型不易分辨。如配製2%凝膠100ml,稱取瓊脂糖2g於三角瓶中,加入100ml 0.5×TBE液,微波爐加熱2-5min,使瓊脂糖完全溶解(注意不要暴沸),置室溫等溫度下降至60℃時,加入終濃度0.5μg/ml的EB液,充分混勻後倒板(注意排除氣體)。
(2)加樣和電泳
上樣時一般取PCR反應液5~10μl,加入3μl溴酚蘭液,充分混勻,加入凝膠加樣孔中。電泳儀可用一般穩壓可調中壓電泳儀,電泳工作液爲0.5×TBE液,接通電源,使樣品由負極向正極移動。60-100V恆壓電泳約30-60分鐘。
(3)檢測
將凝膠板放在紫外透射儀的石英玻璃臺上進行檢測,DNA產物與熒光染料EB形成橙黃色熒光複合物。觀察各泳道是否有橙黃色熒光帶出現,並與擴增時所設的陽性對照比較,判斷陽性或陰性結果。也可在電泳時以標準分子量作對照,判斷其擴增片段是否與設計的大小相一致。
聚丙烯酰胺凝膠電泳比瓊脂糖凝膠電泳繁瑣,但在引物純化、PCR擴增指紋圖、多重PCR擴增、PCR擴增產物的酶切限制性長度多態性分析時常用到。
①分辨率很強,可達1bp;
②能裝載的DNA量大,達每孔10μgDNA;
③回收的DNA純度高;
聚丙烯酰胺凝膠電泳分辨DNA片段的有效範圍:
(1)制膠
在兩塊制膠玻璃板中間放上墊片,放上制膠架,擰緊制膠螺絲夾緊玻璃板。
製備適量合適濃度的凝膠,使之略多於膠板。製備100μl體積凝膠的配製方法見下表。
不同濃度(%)聚丙酰胺凝膠的製法:
試劑 | 3.5% | 5% | 8% | 12% | 20% |
11.6 | 16.6 | 26.6 | 40 | 66.6 | |
水(ml) | 67.7 | 62.7 | 26.6 | 40 | 66.6 |
5×TBE(ml) | 20 | 20 | 20 | 20 | 20 |
10%過硫酸銨(ml) | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 |
TEMED(ml) | 0.035 | 0.035 | 0.035 | 0.035 | 0.035 |
在加入TEMED和10%過硫酸銨後,立即混勻,倒入放置45℃角的玻璃板內,完全注滿(注意不要有氣泡),迅速將玻璃板放置水平位置。立即放成型梳子於腔頂並夾緊,待30-60分鐘膠聚合後,取下膠板,放入電泳槽中固定。
(2)加樣和電泳
上下槽中加入1×TBE電泳緩衝液,溢過加樣孔,拔出梳子,排出加樣孔中的氣泡,將PCR產物或限制性內切酶消化產物2與1上樣緩衝液混勻,用微量注射器加入加樣孔底部,使樣品在孔底成一層,兩側孔中加入標準分子量的DNA Marker。
以2-10V/cm電壓電泳,電泳時間足夠長,使片斷足以分開,待指示劑走到適宜位置時關閉電源,將膠片取出。
(3)銀染
分開兩塊玻璃板,將凝膠片放入100ml銀染固定液中振盪15min,雙蒸水洗3次,每次2min,然後將凝膠片轉入100ml 0.2%的AgNO3中於搖牀上染色約10min,吸去AgNO3液,用雙蒸水洗3次,每次30s,加入100ml顯色液約7-10min,待DNA帶顯示清除時,吸去顯色液,加入固定液Na2CO3,靜置2-3min,取出凝膠觀察。
5.2 PCR產物測序
PCR技術代替了爲測序而反覆進行的分子克隆和模板製備步驟。PCR技術與自動測 序技術相結合後,它將成爲一種最快、最有效的測定核苷權序列的方法。本章主要綜 述各種測模板的製備以及如何完成PCR產物的直接測序。與傳統的將PCR片段克隆入質 粒或病毒基因組相比,直接序列分析有兩個主要的優點:1)由於它是一個不依賴於生 物體(如細菌、病毒等)的體外系統,因而它更容易標準化(即例於自化2);對於每個待 測樣品,只需測定一個單鏈序列,因而它也就更快和更準確。相比之下,間接測序爲 了區別在PCR反應過程中由DNA聚合酶引入的隨機錯朽核苷酸所引起的原始基因組序列 中的突變,以及由體外重組而產生的人工擴增產物,如產生鑲嵌等位基因(混合克隆) 等,每個樣品不得不測定幾個PCR產物克隆的序列。
不需藉助在細菌中克隆就可直接獲得清蜥和準確的序列結果,其難易程度取決 於:1)只擴增靶序列的PCR引物的擴增能力(通常稱爲PCR特異性);2用以獲笪測序模板 的方法。與引物物異性及模板製備有關的問題將在下面談到。雖然DNA測序的化學法 (Maxam-Gilbrt)可以用來直接測序,但我們這裏僅討論Sanger的末端終止法。
最佳的PCR條件
PCR反應的特異性在很大程度上是由寡聚核苷酸引物的序列所決定的。對於特定 的引物組,PCR的特異性可以由於採用最佳的反應條件、退火溫度和PCR緩衝液中的 MgCl2濃度而發生明顯的變化。如果用標準的1.5mM的MgCl2濃度不能得到所需的特異 性PCR產物,我們建議MgCl2的終濃度爲1.4∽2.5mM之間,以0.2mM增長率來改變MgCl2 濃度,以選擇最佳Mg++濃度。一個較常遇見的問題是,使PCR特異性達到最高的MgCl2 濃度導致PCR擴增失敗(dropouts)。這可能是由於模板DNA溶液中有較高的EDTA,它隆 低了提供給Taq DNA聚合酶MgCl2量。因此,對於擴增溶解在TE(1mM EDTA)中的DNA, 我們建議用含2.0mM的MgCl2的PCR緩衝液。
PCR反應的溫度範圍包含有三個不同的恆定期:變性期、退火期和延伸期,還有它 們之間的過渡期。爲了獲得用於序列分析的模板或雜交探針的單鏈DNA,通常並不需 要改變參數。
凝膠純化PCR擴增的靶序列
如果最佳PCR反應條件不能產生所需的特異性產物,可採用新的寡核苷酸重新進 行擴增,或用凝膠電泳來分離不同的PCR產物,而後再各自重新擴增進行序列分析。 長度不同的片段可以用瓊脂糖凝膠電泳來分離:
1.片段大小在80-100bp時,可採用3%NuSieve1%普通瓊脂糖或用聚丙烯酰胺膠來 分離。
2.從膠上切下含所南非PCR片段的薄片。
3.加入50∽100μlTE浸泡膠片,可反覆凍融或放置幾個小時使DNA從膠中擴散出 來。
4.取少量(1-5%)進行第二次擴增,爲得到純淨單一的產物,用於第二次PCR擴增 的凝膠抽提物的量必須少於1ng。
5.現已發現瓊脂糖含有抑制Tab聚合酶活的物質。因而,如需重新擴增供Tab聚合 酶測序用的片段,最好用丙烯酰胺電泳分離。
當用PCR引物擴增幾個同樣長度的相關序列時,如來自幾個新近複製的基因,或 重複序列或保守的信號序列(conserved signal sequences)的同樣顯子,可以通過下 列兩種不同方法來改進電泳分離。1).先用只切割某一模板的內切酶進行酶切,而後 電泳純化完整的PCR產物。2).用可使核苷酸序列不同的擴增產物分開的電泳系統。下 一個部份將討論此類系統中的變性甲酰胺梯度凝膠系統。採用方法一時,必須事先了 解在不同PCR產物中的內切酶位點。
雜合體的直接測序
當兩個等位基因由於單一位點突變而產生差異時,用一個PCR引物直接進行測 序,能找出雜合位點。但是,帶有幾個點突變或短片段的插入/缺失的等位模板用PC R引物之一來直接測序,將產生複合序列帶(compound sequencing ladders)。有四種 方法可以確定幾種點突在型並從雜合體中獲得單個等位基因的序列:1).克隆分離不同 的模板;2).在測序之前,利用模板之間核苷酸序列的差異,用電泳技術分離不同的模 板;3).在測序反應中只啓動一個等位基因;4).只擴增一個等位基因。
測序模板的製備
與PCR產物的直接測序有關的一些問題是變性後由於擴增片段的兩條鏈可以迅速 結合起來,從而阻止了測序引物與它的互補序列退火,或阻止了引物──模板複合物 的延伸。在測序反應中,模板鏈重新結合使一小部份模析驂與測序從而弱化最終的測 序條帶。爲減少此問題,可用改進的標準雙鏈DNA測序法或用PCR製備單鏈模板。
雙鏈DNA模板
有兩種不同方法用來製備測序用的模板,目前都已用來測定共價閉環雙鏈質粒模 板的序列。用這些方法來進行PCR產物的測序通常是較困騅的,因爲短的線性模板比 團環質粒的雙鏈更易於重新結合。在這兩種方法中,PCR片段可以由凝膠電膠或在電 泳前先進行旋轉透析(Spin-dialysis)純化。
1.在室溫下,模板先在0.2M NaOH中變性5分鐘,冰凍,加入0.4倍的5M醋酸銨 (pH7.5)來中和反應。立即用四體積的無水乙醇沉澱DNA。在合適的退火溫度下,加入 測序緩衝引物。
2.在95℃下保溫5分鐘來變性模板,冰浴(或在乾冰乙醇中)快速冷卻離心管,以 減少鏈的重新結合。加入測序引物並使反應達到合適的溫度。測序引物在變性前或後 加入都合適。
單鏈DNA模板
使用單鏈模板可以避免測序中鏈的重新結合。單鏈模板可以從雙鏈DNA模板經鏈 分離凝膠中製備,或用PCR反應制備。大於500bp的單鏈DNA片段,可從瓊脂糖凝膠中 分離笪到,但它不適合於更短的單鏈。製備單鏈DNA的另一種不同方法是在PCR反應中 使用一個生物素標記的引物,變性後的PCR產物經過一個親合素柱而使兩條鏈得到分 離,只有生物素標記的鏈纔可結合到柱上。然而,最簡單的方法是用改進的PCR方 法,用此方法製備既定的單鏈DNA。在這個反應中(不對稱PCR,asymmetric PCR),在 開始的20-25個循環中,兩個比率不對稱的擴增引物產生出雙鏈DNA,當限量的那個引 物耗光後,隨後的5-10個循環就產生出ssDNA。單鏈DNA在約在25個循環後開始出現, 這時限量的引物也幾乎耗盡。經過一個短暫快速上升後,ssDNA就開始如預期的那樣 呈線性積累,這時反應中僅有一個引物存在。各種比率的引物都以這種形式產生 ssDNA。一般對100μlPCR反應體系而言,引物比率爲50pmo:0.5pmol,經過30個循環 後,大約可產生1-3pmol的ssDNA。ssDNA的產量可用下列幾種方法來估計:a)。在PCR 反應中,除了加入正常數量的dDNA外,還加入32P-dNTP。取10%的反應產物在薄的3% NuSieve+1%普通瓊脂糖凝膠中電泳,乾燥並與膠片一起曝光。b).取5%的反應物來走 膠,轉移到膜上,並用與ssDNA互補的寡核苷酸爲探針雜交。ssDNA不能用EB染色來定 量,因爲ssDNA有形成二級結構的趨勢且染料的插入在模板中是隨機的。使用不對稱 引物比例的擴增效率比兩種引物均過量80-90%)的擴增效率低(70%)。在實驗中,這可 通過增加PCR循環的次數來彌補。若不對稱的PCR反應不能產生足夠數量的ssDNA,可 以試一試不同的引物比率;b).再加5-10個PCR循環;c).在最後五個循環中多加Tab聚合 酶(2U);d).試一下相反的不對稱引物比率。有時,相反的不對稱引物比率可能給出不 同產量的ssDNA。製備出的單鏈DNA用限量的那個PCR引物或用一個內引物來測序,並 應用常規方法進行摻入測序(incorporation sequencing)或標記引物測序。製備出的 ssDNA鏈可能有一個不連續的5'-末端,但可能在靠近3'-末端不同位點處被切去,這 是由於延伸過程中終止過早所產生的。然而,對於測序反應中所使用的任何一種引 物,只有完整的ssDNA可以用作測序模板。
最近還報道了另一種方法製備單鏈核酸模板進行直接測序。這種方法包括在一個 PCR引物上加入噬菌體的啓動子,轉錄出該PCR產物的RNA拷貝,再用反轉錄酶不測 序。但由於擴增反應後附加了一個酶促反應步驟和限於用反轉錄酶作爲測序酶,這種 方法的應用受到很大的限制。
用不耐熱DNA聚合酶進行PCR產物的測序
一些不耐熱DNA聚合酶已經用來直接測序體外擴增的DNA。使用Klenow聚合酶,修 飾T7DNA聚合酶(測序酶)及反轉錄酶來測序。
用Taq聚合酶進行PCR產物的測序
Tab聚合酶除了用作PCR擴增外,它還是DNA測序的理想的酶。Tab聚合酶具有高度 的延伸能力(processivity),較高的摻入速率,並可用一些核酶類似物如7-Deaza-2' -脫氧鳥苷-5'-三磷酸來解決DNA測序中的壓縮問題。除了這些與T7DNA聚合酶相同的 性質外,它有較高的熱穩定性,使反應溫度可達55-70℃,這可使大量的二級結構解 體。可是,對於dNTPs而言,它有一個相對高的Km值(≈10-20μM)並缺乏3'-外切酶活 性。當dNTP濃度低於1μM時,它將產生錯配和不正確的終止。瓊脂和瓊脂糖中都有 Tab聚合酶的抑制物,但這些可通過增加測序反應中Tab聚合酶的量(2U-10U)得到部分 解決。當測定克隆插入子的PCR產物序列時時,由噬菌體液裂解製備的靶基因中並不 含有從平板溶解出來的抑制物。
對帶有強二級結構的DNA進行測序
帶有強二級結構的DNA測序可能產生兩個問題:1).由於Tab聚合酶不能完全延伸的 模板頻率很高,因而降低了PCR的效率,2.).在測序反應中,壓縮被測DNA序列的長 度。
研究結果表明:在PCR中使用Tab聚合酶(50-70℃)的高反應溫度足夠解決大部份短 的二級結構。但強的二級結構會產生強烈的抑制作用。這些只能通過加入與dGTP比例 事適的鹼基類似物c7dGTP後,纔可解決。同樣的鹼基類似物也可用在測序反應中,來 解決壓縮問題(compression problem)。Tab聚合酶可以有效地摻入c7dGTP,但不能用 次黃嘌吟核苷作爲鹼基類似物。同樣的鹼基類似物也可用在測序反應中,來解決壓縮 問題(COMPRESSION)。Tab聚合酶可以有效地摻入c7dGTP,但不能用次黃嘌呤核苷作 爲鹼基類似物。
PCR擴增DNA測序中涉及到的錯誤
在原始靶基因序列和由它擴增出的PCR產物之間可能有兩種不一致情況:1).由點 突變引起的差異;2).由體外重組的等位基因引起的差異。點突變引起的差異是由於 每”個循環中每個核苷酸大約有2×10-4機率發生錯誤摻入。經過30個循環擴增後,每 個PCR產物平均在每400-4000個鹼基對內存有差異。這個錯誤摻入的機率有時比 Klenow聚合酶(8×10-5)更高。體外重組產生的等位基因(shuffle clone)j是鑲嵌PCR 產物,這可能是在以後的循環中由部分延伸的PCR產物造成的,因爲它可作爲另一個 等位基因模板的引物。由於酶量不足以用來延伸所有的模板,使得在以後的反應循環 中主要積累這些等位基因。除非雜合體中的兩個等位基因在其PCR引物上有幾個不同 位點的差異,這些人工產物進不能檢測出來的。
當用PCR產物來克隆、測序並從一組相同克隆子序列(consensus sequence)推斷 出各個等位基因的序列時,就必須考慮到這兩種不同類型的錯誤。相比之下,當PCR 產物用來直接測序時,由錯誤摻入或鑲嵌而產生的錯誤PCR產物並不影響PCR測序。在 各個PCR產物中由點突變引起的差異與相同序列比是檢測不出的。既使這個突變(如錯 誤摻入)在以一個DNA分子爲模板的如單個精子DNA擴增反應的第一個循環中產生,它 也僅佔該位點正常核苷酸的25%。對起始DNA模板不止一個的擴增反應,在任一位點上 含有一個錯配核苷酸的PCR產物的頻率低到檢測不出的程度。雖然目前並不很瞭解鑲 嵌基因形成的條件,但相信這些等位基因是在反應最後幾個循環中積累起來的,因爲 這時酶量可能不足以用來延伸所有的模板。除非序列中有非常強烈的二級結構,在某 一特定位點上使鏈延伸終止,結果產生一個單一的人工基因。鑲嵌基因不會影響正常 序列的測定。
測序反應的自動化
DNA測序的可重複性使它適合於完全或部分自動化。已有用常規的放射性標記引 物,或熒光終止標記或測序引物進行自動化測序的一系列儀器。但是,這些儀器僅使 分析的前半部分自動化,凝膠電泳、讀帶和將序列輸到計算機中用後半部分自動化來 補足。前半部有兩個階段:模板的製備測序模反應,可很容易地進行自動化反應。用 Tab酶進行PCR擴增和直接測序是製備測序模板和測序反應終產物的最有效方法。一旦 使用自動進樣器,就可進行自動化反應,僅電泳分析由人工進行。這對許多一直使用 人工電泳測序反應的實驗室來說,都將從中受益。
5.3 PCR產物克隆方法
平端連接
通常情況下,PCR產物可直接與平端載體DNA進行連接,但其連接效 率效低。因爲TaqDNA聚合酶具有非模板依賴性末端轉移酶活性,能 在兩6條DNA鏈的3'末端加上一個多餘的鹼基,使合成的PCR產物成爲 3'突出一個鹼基的DNA分子。這種DNA分子的連接效率很低。由於PCR 產物的效率通過較高,。在採用大量T4DNA連接酶並配以5—10u T4 RNA連接酶時,可顯著提高其連接效率。對於較短PCR產物,用PUS19 的HincⅡ位點進行克隆,以X-gal和IPTG篩選,常可得到足量重組 子。另一種提高克隆效率的途徑是先用Klenow大片段或T4DNA聚合酶 消去3'末端突出鹼基將PCR產物變成平端DNA,然後再用平端連接法 克隆PCR產物。
粘端連接
引物中設計入限制酶位點:由於PCR引物的5'末端可以增加一些非 互補鹼基,因此可以在兩引物的5'末端設計單限制酶或雙限制酶切 位點。這樣得到的PCR產物用限制酶消化產生粘性末端,即可與有互 補粘端的載體DNA重組。這種克隆方法效率較高,且當兩引物中設計 不同酶切位點時,可有效地定向克隆PCR產物。其缺點是需要加長 PCR引物,除限制酶識別序列外,還需要在其5'端多合成3—4個鹼基 以利於限制性內切酶與PCR產物末端的穩定結合。即使如此,其酶切 效率也不夠高。其中尤以NotI、XhoI和XbaI等較爲難切。採用突變 PCR方法可克服上述缺點。該方法是通過在兩PCR引物序列中改變1至 數個核苷酸創造出一個限制性內切酶位點。鑑於PCR引物的3'末端序 列的互補性是PCR成功的關鍵,在PCR引物的中部或近5'端改變1個或 幾個鹼基對PCR擴增效果影響不大。這種方法不需要增加PCR引物的 長度,而且酶切效果優於5'加端法。對於特定DNA片段的克隆,此方 法較爲經濟、實用。但對於基因診斷PCR產物的克隆,似乎5'加端法 更爲適宜。
T4DNA聚合回切產生粘端
如PCR兩引物的5'末端是A或T,則可在 其5'端分別加上CG和CCGG。用此二引物擴增的PCR產物在dATP和dTTP 存在的情況下,用T4DNA聚合酶進行處理,則T4DNA聚合酶因具有3'→ 5'外切酶活性而消去3'末端的G和C,產生AccI和XmaI粘性末端(圖1)。 此DNA片段直接與用AccI和XmaI切開的載體進行連接。這種方法只需 在PCR引物的5'端加2—4個鹼基,但其可選擇的限制酶類有限。
T-vector法
TaqDNA聚合酶能在平端雙鏈DNA的3'末端加一個鹼 基,所加鹼基幾乎全是腺苷。據此,Marchuk等人採用3'端突出一個 胸苷的質粒DNA來克隆PCR產物,其克隆效率比平端的連接至少高出 100倍。他們用EcoRV將pBluescript切成平端,然後在2mmol/LdTTP 存在下,用TaqDNA聚合酶催化pBluescript的兩個3'末端各加一處胸 苷。因爲在4種dNTP都存在時,Taq聚合酶選擇性參入dATP,而當僅 一種dNTP存在時,它只能參入該種鹼基。因此,在只加入ddTTP時, 用TaqDNA聚合酶可使平端載體DNA轉變成3'末端突出一個胸苷的T尾 載體,稱爲T-vector。用這種T-vectorsk可以較有效地直接克隆 PCR產物。Hotton等人也報道了另一種製備T-vector的方法。他們 使用脫氧核苷酸末端轉移酶在切成平端的載體DNA的3'末端加上一個 胸苷。由於末端轉移酶可以催化多個鹼基(ddTTP)作爲底物,使平 端載體DNA分子的兩個3'末端各加上一個T。用這種方法製備的T-vector 的不同之處在於前者3'末端不能與待克隆PCR產物的5'末端連接,僅 5'末端可與PCR產物的3'末端形成磷酸二脂鍵。
共環消解法:最近,Jung等人報道了一種有效的PCR產物克隆方 法。用磷酸化的PCR引物擴增得到的PCR產物,先用T4DNA連接酶催化 連接反應,使5'端帶有限制酶切位點的擴增DNA片段連接成共環結 構。然後再用相應的限制酶進行消化,產生粘端DNA片段。對於對稱 性限制酶位點,只需在引蛾的5'末端加上一關識別序列,因爲在串 接成共環後能恢復限制酶切位點難於切開的缺點,且可用於雙限制 酶切位點的設計,只不過有PCR產物共環化後,僅約1/4的限制酶切 點得以恢復。故此法較適用於單限制酶位點的克隆。
無連接酶亞克隆法(A)
無連接酶克隆法(ligase-free subcloning,LFS)是利用引物5’ 末端附加鹼基修飾法,修飾鹼基不是酶切位點,而是與某一質粒兩 端分別互補的鹼基。兩引物的3’端約20—25個核苷酸分別與待擴增 DNA兩翼互補,5’端各有約24個核苷酸分別與線性化質粒的3’端相 同的附加序列。由於線性化質粒的3’端序列各不相同,PCR片段可 以通過選擇各引物的合適5’附加序列與引物3’端定向雜交。
由此物a和b產生的兩端有附加序列的PCR產物與未反應引物分離後, 分別加入兩隻含有線性化質粒的反應管中進行第二次PCR。第1管中 用引物a和c,引物a即爲第一PCR擴增的上游引物a,引物c爲下游引 物,與緊鄰5’端附加序列內測的質粒(+)鏈互補。同樣,第2管的 引物爲b和d,引物b與第一次PCR擴增的下游引物b相同,引物d爲上 遊引物,與緊鄰5’附加序列內側的質粒(-)鏈互補。
第二次PCR的第一循環中,PCR產物與質粒均變性與復性。除自身復 性產物(這種復性產物不被擴增)外,PCR產物與質粒可通過各自3’ 端互補序列雜交成部分異源雙鏈,延伸時,重疊的3’端互爲此物沿 各自互補鏈延伸,結果可產生PCR片段與線性質粒的“連接”。然後 兩管中PCR擴增各進行15—20個循環。這便可產生大量一端管1)或 另一端連接有PCR插入片段的質粒。
第二次PCR後,將第1管與第2管反應液混合,用鹼變性雙鏈,中和後 稀釋變性的DNA。反應管中的單鏈DNA可以復性或幾種不同的產物, 除各自本身復性產物外,管1產物ssDNA與管2中ssDNA可形成部分異 源雙鏈DNA,並各自有一較長的5’或3’懸端,這種長的5’或3’懸 端相互互補,在低DNA濃度時可復性產生環化DNA。
儘管這種環化的DNA有兩個缺口,但它們可以直接用來轉化受體大腸 直桿菌。一旦進入體內,兩個缺口便共價連接,修復的質粒即可復 制,下面以從λ噬菌體DNA中擴增-500bp片段,並克隆入pGem4Z載體 中爲例說明LFS法。
這種方法同樣適於複雜基因組中基因片段的克隆。需注意的是第一 次PCR時兩引物的5’附加序列不應太短以免影響第二次PCR時異源雙 鏈的形成,以24個核苷酸較爲合適。擴增時若形成,引物二聚體, 一定要去除,否則會嚴重影響轉化率。用LFS法已成功地克隆了長達 1.7kb的基因片段。這種方法的優點是:①可用於常規方法無法進行 亞克隆的片段;②適於任何PCR產物和任何質粒;③可亞克隆特殊目 的(如含點突變、缺失或插入等)片段;④在某些情況下,對已構 建了啓動子或增強子等序列的載體,可使待表達片段插入定向合適 位置;⑤較快,可在1d內完成,較常規方法可靠,不需DNA連接酶。
DNA克隆是分子生物學的重要內容。特定基因的克隆常因兩端缺乏合 適限制酶切點而受因,cDNA的克隆通常也效率不高、篩選因難。採 用PCR技術行DNA和cDNA的克隆,則可大大縮短克隆時間,比之全基 因合成更爲經濟和方便,因而愈來愈受重視。用PCR方法進行傳染性 疾病和遺傳性疾病的診斷常遇到產物的異性問題和分型問題,採用 產物克隆和測序方法,比之寡核苷酸探針雜交方法更爲準確。隨着 PCR技術的不斷髮展和推廣,新的PCR產物的克隆方法也將不斷出現。
6 參考資料
- ^ [1] 張新宇,高燕寧."PCR引物設計及軟件使用技巧".中國協和醫科大學中國醫學科學院腫瘤研究所