2 附錄Ⅵ A 氮測定法(半微量法)
本法系依據含氮有機物經硫酸消化後,生成硫酸銨,硫酸銨被氫氧化鈉分解釋放出氨,後者借水蒸氣被蒸餾入硼酸液中生成硼酸銨,最後用強酸滴定,依據強酸消耗量可計算出供試品的氮含量。
2.1 試劑
(1)消化劑 稱取硫酸銅(CuSO4·5H2O10g、硫酸鉀100g,置乳鉢內,共同研細,混勻。
(2)混合指示液 0.2%溴甲酚綠乙醇溶液5份與0.1%甲基紅乙醇溶液2份混合配成。
(3)2%硼酸吸收液 稱取硼酸20g,加水溶解並稀釋至1000ml,加混合指示液10ml,混勻。
(4)硫酸滴定液(0.05mol/L) 量取硫酸3ml,緩緩注入適量水中,冷卻至室溫,加水稀釋至1000ml,搖勻。
取在270~300℃乾燥至恆重的基準無水碳酸鈉約0.15g,精密稱定,加水50ml使溶解,加甲基紅-溴甲酚綠混合指示劑10滴,用本液滴定至溶液由綠色轉變爲紫紅色時,煮沸2分鐘,冷卻至室溫,繼續滴定至溶液由綠色變爲暗紫色。每1ml硫酸滴定液(0.05mol/L)相當於5.30mg的無水碳酸鈉。根據本液的消耗量與無水碳酸鈉的取用量,算出本液的濃度,即得。
(5)硫酸滴定液(0.005mol/L) 精密量取硫酸滴定液(0.05mol/L) 100ml,置1000ml量瓶中,加水稀釋至刻度,搖勻。
2.2 測定法
精密量取一定體積的供試品(約相當於含氮量1.0~2.0mg),置凱氏定氮瓶中,加消化劑約0.3g,硫酸1ml消化至澄明,呈藍綠色,繼續消化約60分鐘。量取2%硼酸吸收液10ml置100ml錐形瓶內,將凱氏蒸餾器冷凝管末端浸入硼酸吸收液內,將消化好的供試品移入凱氏蒸餾器內,用水洗定氮瓶3~4次,將洗液移人蒸餾器內,再加入50%氫氧化鈉溶液5ml,然後進行蒸餾,待接收液總體積約35~50ml,將冷凝管末端移出液麪,使蒸汽繼續沖洗約1分鐘,用水淋洗尖端後停止蒸餾。接收液用硫酸滴定液(0.005mol/L)進行滴定,至溶液由藍綠色變爲灰紫色,並將滴定的結果用空白試驗校正。
按下式計算:
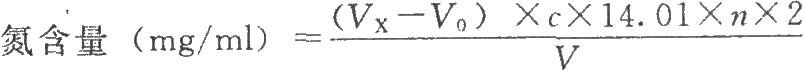
式中VX爲供試品消耗酸滴定液的體積,ml;
n爲供試品的稀釋倍數;
V爲供試品溶液的體積,ml;
14.01爲氮的相對原子質量。
2.3 【附註】
(1)蒸餾前應蒸洗蒸餾器15分鐘以上。
(3)也可用全自動凱氏定氮儀測定,按儀器使用說明書進行操作。
3 附錄Ⅵ B 蛋白質測定法
3.1 第一法 凱氏定氮法
3.1.1 1.鎢酸沉澱法
本法系通過測定供試品的總氮含量以及經鎢酸沉澱去除蛋白質的供試品濾液中的非蛋白氮含量,計算出蛋白質的含量。
供試品溶液的製備
(1)總氮測定溶液的製備 精密量取供試品1ml,用生理氯化鈉溶液準確稀釋至每1ml含氮量約1mg。
(2)非蛋白氮測定溶液的製備 精密量取供試品2ml,加水14ml、10%鎢酸鈉溶液2ml、硫酸溶液(1.86→100) 2ml,搖勻,靜置30分鐘過濾,取濾液測定。
測定法 精密量取總氮測定溶液1ml,置凱氏定氮瓶中,照氮測定法(2010年版藥典三部附錄Ⅵ A)測定供試品中總氮含量,同時做空白對照。
精密量取非蛋白氮測定溶液5ml,置凱氏定氮瓶中,照氮測定法(2010年版藥典三部附錄Ⅵ A)測定供試品中非蛋白氮含量。按下式計算:

式中CTN爲供試品溶液總氮含量,mg/ml;
CNPN爲供試品溶液非蛋白氮含量,mg/ml;
V0爲空白試驗消耗酸漓定液的體積,ml;
CPN爲供試品蛋白氮含量,mg/ml;
n爲供試品的稀釋倍數;
V1爲供試品總氮測定溶液的體積,ml;
V2爲供試品非氮測定溶液的體積,ml;
14.01爲氮的相對原予質量;
【附註】
(1)供試品蛋白質含量如超過10%(g/ml),除蛋白質時應適當加大供試品稀釋倍數,10%鎢酸鈉溶液及硫酸溶液用量相應地按比例增加,使溶液中的鎢酸濃度仍保持1%。
(2)總氮測定和非蛋白氮測定可用同一空白對照。
3.1.2 2.三氯乙酸沉澱法
本法系將供試品經三氯乙酸沉澱,通過測定該沉澱中的蛋白氮含量,計算出蛋白質的含量。
測定法 精確量取適宜體積的供試品(每1ml含蛋白質4~10mg)於適宜的尖底離心管中,加等體積的12%三氯乙酸(12→100)混勻,靜置30分鐘後,以每分鐘4000轉離心,棄上清液,用約3ml水分數次將沉澱洗入凱氏定氮瓶中,照氮測定法(2010年版藥典三部附錄Ⅵ A)測定供試品中總氮含量,同時做空白對照。
按下式計算:
供試品蛋白質含量(mg/ml) 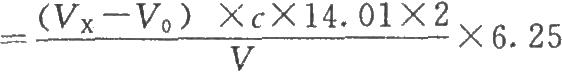
式中VX爲供試品消耗酸滴定液的體積,ml;
V爲供試品的體積,ml。
14.01爲氮的相對原子質量;
3.2 第二法 Lowry法
本法用於微量蛋白質的含量測定。蛋白質在鹼性溶液中可形成銅-蛋白質複合物,此複合物加入酚試劑後,產生藍色化合物,該藍色化合物在波長650nm處的吸光度與蛋白質含量成正比,根據供試品的吸光度,計算供試品的蛋白質含量。
3.2.1 試劑
(1)酚試劑 稱取鎢酸鈉(Na2WO4·2H2O) 100g、鉬酸鈉(Na2MnO4·2H2O) 25g,置1500ml蒸餾瓶中,加入700ml水、85%磷酸50ml、鹽酸100ml,上連回流管(使用木塞或錫紙包裹的橡皮塞)微沸迴流10小時。取下回流管,加入硫酸鋰150g、水50ml、溴液幾滴,煮沸約15分鐘,驅除過量的溴。冷卻,加水至1000ml,過濾,爲酚試劑貯備液。
酚試劑貯備液用氫氧化鈉滴定液(0.5mol/L)測定酸濃度,然後加水稀釋至相當於1mol/L鹽酸濃度。
(2)鹼性銅溶液 量取0.1mol/L酒石酸鉀溶液與0.04mol/L硫酸銅溶液各0.5ml,4%碳酸鈉溶液與0.8%氫氧化鈉溶液各25ml,搖勻,即得。本液應臨用配製。
(3)氫氧化鈉滴定液(0.5mol/L)的配製及標定取氫氧化鈉適量,加水攪拌使溶解成飽和溶液,冷卻後,置聚乙烯塑料瓶中,靜置數日,澄清後,量取澄清的氫氧化鈉飽和溶液28ml,加新煮沸後冷卻的水,使成1000ml,搖勻。
取在105℃乾燥至恆重的基準鄰苯二甲酸氫鉀約3g,精密稱定,加新沸過的冷水50ml,振搖,使其儘量溶解;加酚酞指示劑2滴,用本液滴定;在接近終點時,應使鄰苯二甲酸氫鉀完全溶解,滴定至溶液顯粉紅色。每1ml氫氧化鈉滴定液相當於102.1mg的鄰苯二甲酸氫鉀。根據本液的消耗量與鄰苯二甲酸氫鉀的取用量,算出本液的濃度。
3.2.2 標準蛋白質溶液的製備
用水將蛋白質標準品定量稀釋至每1ml含1mg,作爲貯備液。精密量取貯備液2.5ml置25ml量瓶中,用水稀釋至刻度,搖勻,即爲每1ml含100μg的標準蛋白質溶液。
3.2.3 測定法
精密量取一定體積供試品(含蛋白質50μg左右)置試管內,加水至1ml,加鹼性銅溶液5ml,搖勻,室溫放置10分鐘,快速加入酚試劑0.5ml,搖勻,室溫放置30分鐘,顯色後,照紫外一可見分光光度法(2010年版藥典三部附錄Ⅱ A),在波長650nm處測定吸光度(呈色後,如發現渾濁,經每分鐘3000轉離心15分鐘後,取上清液測定)。精密量取標準蛋白質溶液0.2ml、0.4ml、0.6ml、0.8ml、1.0ml分別置於試管中,自“加水至1ml”起,同法操作。準確量取水1ml,自“加鹼性銅溶液5ml”起,同法操作,作空白對照。以標準品的蛋白質濃度對其相應吸光度作直線迴歸,求得直線迴歸方程;將測得供試品的吸光度代入直線迴歸方程,即得供試品的蛋白質含量。
3.3 第三法 雙縮脲法
本法系依據蛋白質肽鍵在鹼性溶液中與Cu2+形成紫紅色絡合物,其顏色深淺與蛋白質含量成正比,利用標準蛋白質溶液作對照,採用紫外一可見分光光度法測定供試品蛋白質含量。
3.3.1 試劑
雙縮脲試劑 取硫酸銅(CuSO4·5H2O)3.0g、酒石酸鉀鈉(KNaC4H4O6·4H2O)9.0g、碘化鉀5.0g、氫氧化鈉24g,加水溶解並稀釋至1000ml,搖勻,即得。
3.3.2 標準蛋白質溶液的製備
3.3.3 供試品溶液的製備
精密量取適量的供試品,加水製成每1ml約含50mg的溶液。
3.3.4 測定法
分別精密量取供試品溶液與標準蛋白質溶液各0.05ml置玻璃試管中,分別加雙縮脲試劑4.0ml,混勻,置37℃水浴中30分鐘,照紫外-可見分光光度法(2010年版藥典三部附錄Ⅱ A),在波長540nm處測定吸光度。另精密量取水0.05ml,自“加雙縮脲試劑4.0ml”起,同法操作,作爲空白對照。
按下式計算:
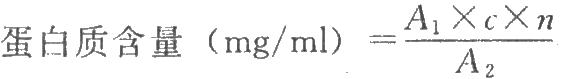
式中A1爲供試品溶液的吸光度;
n爲供試品稀釋倍數。
【附註】 本法的測定範圍爲1~10mg。
4 附錄Ⅵ C 唾液酸測定法(間苯二酚顯色法)
本法系用酸水解方法將結合狀態的唾液酸變成遊離狀態,遊離狀態的唾液酸與間苯二酚反應生成有色化合物,再用有機酸萃取後,測定唾液酸含量。
4.1 唾液酸對照品溶液(200Vg/ml)的製備
精密稱取唾液酸對照品10.52mg(1μg唾液酸相當於3.24nmol),置10ml量瓶中,加水溶解並稀釋至刻度,混勻,即爲唾液酸貯備液(1mg/ml),按一次使用量分裝,-70℃貯存,有效期1年。僅可凍融1次。4℃保存使用期爲2周。精密量取唾液酸貯備液1ml,置5ml量瓶中,加水至刻度,即爲每1ml含200μg的唾液酸對照品溶液,用前配製。
4.2 測定法
取供試品適量,加水稀釋至蛋白質濃度約爲每1ml含0.2~0.4mg,作爲供試品溶液。按下表取唾液酸對照品溶液、水及供試品溶液於10ml玻璃試管中,混勻,每管再加入間苯二酚-鹽酸溶液(分別量取2%間苯二酚溶液2.5ml、0.1mol/L硫酸銅溶液62.5μl、25%鹽酸溶液20ml,加水稀釋至25ml,混勻。試驗前4小時內配製)1ml,,加蓋,沸水煮沸30分鐘(水浴面高於液麪約2cm),取出置冰浴中3分鐘(同時振搖)後,每管加乙酸丁酯-丁醇液(取4體積乙酸丁酯與1體積丁醇混勻,室溫下保存,12小時內使用)2ml,充分混勻,室溫放置10分鐘,照紫外-可見分光光度法(2010年版藥典三部附錄Ⅱ A),在波長580nm處測定吸光度。
以唾液酸對照品溶液的濃度對其相應的吸光度作直線迴歸(相關係數應不低於0.99),由直線迴歸方程求出5μg唾液酸的吸光度,再按下式計算:
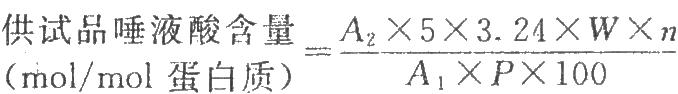
式中A1爲5μg唾液酸的吸光度;
A2爲供試品的吸光度;
n爲供試品稀釋倍數;
P爲供試品蛋白質含量,μg/μl;
W爲1nmol促紅素的量[1],相當於30.6μg。
5 附錄Ⅵ D 乙醇殘留量測定法(康衛皿擴散法)
本法系依據乙醇在飽和碳酸鈉溶液中加熱逸出,被重鉻酸鉀-硫酸溶液吸收後呈黃綠色至綠色,用比色法測定血液製品中乙醇殘留量。
5.1 測定法
在康衛皿外圈的凸出部位均勻塗抹凡士林,準確量取重鉻酸鉀-硫酸溶液(稱取重鉻酸鉀3.7g,加水150ml,充分溶解後緩慢加入硫酸280ml,放冷,加水至500ml,搖勻)2.0ml加入內圈中,量取飽和碳酸鈉溶液[稱取碳酸鈉(Na2CO3·10H2O)適量,加等重量的水,充分搖勻,取上清液] 1.5ml和精密量取的供試品溶液1.5ml,加入外圈中,立即加蓋玻璃板(粗糙面向下)密封擴散皿,搖勻,80℃反應30分鐘後,取內圈溶液,照紫外-可見分光光度法(2010年版藥典三部附錄Ⅱ A),在波長650nm處測定吸光度(A1)。精密量取無水乙醇適量,加水製成每1ml中含乙醇0.25mg的溶液,即爲對照品溶液。精密量取對照品溶液1.5ml替代供試品,同法操作,測定吸光度(A2)。A1不得大於A2。
6 附錄Ⅵ E 組胺人免疫球蛋白中游離磷酸組胺測定法
本法系依據磷酸組胺與鄰苯二甲醛在鹼性條件下生成熒光衍生物,以此測定組胺人免疫球蛋白中游離磷酸組胺含量。
6.1 磷酸組胺對照品溶液的製備
取磷酸組胺對照品7mg,精密稱定,置25ml量瓶中,用0.1mol/L鹽酸溶液溶解並稀釋至刻度,搖勻,作爲磷酸組胺貯備液,-20℃貯存備用。試驗當天準確量取磷酸組胺貯備液0.1ml,置100ml量瓶中,用0.1mol/L鹽酸溶液稀釋至刻度,即爲磷酸組胺對照品溶液。
6.2 供試品溶液的製備
量取供試品0.5ml,加水1.2ml,混勻,加25%三氯乙酸溶液0.3ml,混勻,以每分鐘4000轉離心10分鐘,取上清液,即爲供試品溶液。
6.3 測定法
量取供試品溶液1.6ml置試管中,加氯化鈉1.5g,再加正丁醇4.0ml、2.5mol/L氫氧化鈉溶液0.2ml,立即混勻5分鐘,靜置後,取出正丁醇相3.6ml加到已裝有0.1mol/L鹽酸溶液1.2ml和正庚烷2.0ml的試管內,振盪5分鐘,棄有機相,量取鹽酸相1.0ml,加入等體積水,再加0.4mol/L氫氧化鈉溶液0.5ml,混勻並迅速加入0.1%鄰苯二甲醛-甲醇溶液0.1ml,立即混勻,置21~22℃10分鐘,加0.5mol/L鹽酸溶液0.5ml終止反應,取終止反應後的溶液200μl,加入酶標板孔中,用熒光酶標儀,在激發波長350nm和發射波長450nm處測熒光強度。
準確量取磷酸組胺對照品溶液1.0ml、0.8ml、0.6ml、0.4ml、0.2ml、0.1ml、0.05ml、0.025ml,分別置試管中,各以0.1mol/L鹽酸溶液補足至1.0ml;向各管中加水0.5ml、25%三氯乙酸溶液0.1ml,混勻,加氯化鈉1.5g,自“加正丁醇4.0ml”起,同法操作。
以磷酸組胺對照品溶液的濃度對其相應的熒光強度作直線迴歸,將供試品溶液的熒光強度代入直線迴歸方程,求出供試晶溶液鹼基含量(G),按下式計算:
供試品遊離磷酸組胺含量(ng/ml)=G×2.76×2.5
【附註】 磷酸組胺分子量爲307.148,對照品溶液濃度按鹼基計,鹼基與磷酸組胺分子量比爲1:2.76。式中2.5爲供試品稀釋倍數。
7 附錄Ⅵ F O-乙酰基測定法
7.1 試劑
(1) 2mol/L鹽酸羥胺溶液 稱取鹽酸羥胺13.9g,加水溶解並稀釋至100ml,冷處保存。
(2)3.5mol/L氫氧化鈉溶液 稱取氫氧化鈉14.0g,加水使溶解並稀釋至100ml。
(3) 4mol/L鹽酸溶液 量取鹽酸33.3ml,加水稀釋至100ml的溶液。
(4)0.37mol/L三氯化鐵一鹽酸溶液 稱取三氯化鐵(FeCl3·6H20) 10.Og,加0.1mol/L鹽酸溶液溶解並稀釋至100ml。
(5)鹼性羥胺溶液 量取等體積的鹽酸羥胺溶液(2mol/L)與氫氧化鈉溶液(3.5mol/L)混合。3小時內使用。
7.2 對照品溶液的製備
精密稱取已乾燥至恆重的氯化乙酰膽鹼22.7mg(或溴化乙酰膽鹼28.3mg),置50ml量瓶中,加0.001mol/L醋酸鈉溶液(pH4.5)溶解並稀釋至刻度,搖勻。
7.3 供試品溶液的製備
取供試品,用水稀釋成O-乙酰基濃度爲0.5~2.5mmol/L的溶液。
7.4 測定法
精密量取氯化乙酰膽鹼(或溴化乙酰膽鹼)對照品溶液0.2ml、0.4ml、0.6ml、0.8ml、1.0ml,分別置試管中,補加水至1ml,加新鮮配製的鹼性羥胺溶液2ml,搖勻,於室溫放置4分鐘,加4mol/L鹽酸1ml,調pH值至1.2±0.2,搖勻,加0.37moI/L三氯化鐵-鹽酸溶液1ml,搖勻,照紫外-可見分光光度法(2010年版藥典三部附錄Ⅱ A),在波長540nm處測定吸光度。另精密量取上述相應的系列對照品溶液,自“補加水至1ml”起,除加酸與加鹼性羥胺的次序顛倒外,同法操作,用作對應的空白對照。
精密量取供試品溶液1ml置試管中,自“加新鮮配製的鹼性羥胺溶液2ml”起,同法操作;另取供試品溶液1ml,與對照品溶液的空白對照同法操作,用作供試品的空白對照。
將標準管各吸光度分別減去相應的空白對照管的吸光度,以標準管中所含的對照品溶液的體積對其相應的吸光度作直線迴歸,將供試品的吸光度減去相應的空白對照管的吸光度後代入直線迴歸方程,計算出每1ml供試品相當於對照品溶液的體積(V,ml)。
供試品中O-乙酰基含量(mmol/L)=V×2.5
8 附錄Ⅵ G 聚乙二醇殘留量測定法
本法系依據聚乙二醇與鋇離子和碘離子形成複合物(1:1).用比色法測定聚乙二醇含量。
8.1 測定法
取供試品適量,用水稀釋,使蛋白質濃度不高於1%,即爲供試品溶液。精密量取供試品溶液1.0ml,加入0.5mol/L高氯酸溶液5.0ml,混勻,室溫放置15分鐘,以每分鐘4000轉離心10分鐘。取上清液4ml,加入氯化鋇溶液(稱取氯化鋇5g,加水溶解至100ml)1.0ml和0.1mol/L碘溶液(稱取碘化鉀2.0g,加少量水溶解,然後加碘1.3g,再加水至50ml,搖勻)0.5ml,混勻,室溫反應15分鐘,照紫外-可見分光光度法(2010年版藥典三部附錄Ⅱ A),在波長535nm處測定吸光度;同時以1ml水代替供試品溶液,同法操作,即爲空白對照。
另精密稱取聚乙二醇(分子量4000或6000)適量,加水溶解,並製成每1ml含聚乙二醇100μg的溶液,即爲聚乙二醇對照品貯備液。
取按下表製備的每1ml含10~50μg的聚乙二醇對照品溶液1.0ml,加入0.5mol/L高氯酸溶液5.0ml,混勻,自“室溫放置15分鐘”起,同法操作。
以聚乙二醇對照品溶液的濃度(μg/ml)對其相應的吸光度作直線迴歸,將供試品溶液吸光度代入直線迴歸方程,計算出供試品溶液聚乙二醇含量F(μg/ml)。
按下式計算:
供試品聚乙二醇含量(g/L)=F×n×10-3
n爲供試品稀釋倍數。
8.2 【附註】
(1)整個比色過程應在試劑加入後的15~45分鐘內完成,否則將要影響結果。
9 附錄Ⅵ H 聚山梨酯80殘留量測定法
本法系依據聚山梨酯80中的聚乙氧基(Polyethoxylated)和銨鈷硫氰酸鹽反應形成藍色復合物,可溶於二氯甲烷,用比色法測定聚山梨酯80含量。
9.1 測定法
量取供試品1.0ml於離心管中,加乙醇-氯化鈉飽和溶液5ml,搖勻,以每分鐘3000轉離心10分鐘,取上清液,再用乙醇-氯化鈉飽和溶液1.0ml小心沖洗管壁,洗液與上清液合併,以每分鐘3000轉離心10分鐘,上清液置55℃水浴中,用空氣吹掃法將其濃縮至0.1~0.5ml,加1ml水溶解。準確加入二氯甲烷2.0ml、硫氰鈷銨溶液(稱取硝酸鈷6.0g、硫氰酸銨40.0g,加水溶解並稀釋至200ml)3.0ml,加塞,混勻,室溫放置1.5小時,每15分鐘振盪1次,測定前靜置半小時,棄上層液,照紫外-可見分光光度法(2010年版藥典三部附錄Ⅱ A),在波長620nm處測定下層二氯甲烷液的吸光度。用二氯甲烷作空白對照。
精密量取聚山梨酯80對照品溶液(取聚山梨酯80約100mg,精密稱定,加水溶解後置100ml量瓶中,加水稀釋至刻度)0μl、10μl、25μl、50μl、75μl、100μl,加入預先加入1ml水的離心管中混勻,準確加入二氯甲烷2.0ml、硫氰鈷銨溶液3.0ml,加塞,混勻,自“室溫放置1.5小時”起,同法操作。
以上述聚山梨酯80系列濃度(μg/ml)對其相應的吸光度作直線迴歸,相關係數應不低於0.98,將供試品吸光度代入直線迴歸方程,求得供試品聚山梨酯80含量(μg/ml)
10 附錄Ⅵ I 戊二醛殘留量測定法
本法系依據戊二醛與2,4-二硝基苯肼反應生成正戊醛二硝基苯肼,用高效液相色譜法,測定供試品中戊二醛含量。
照高效液相色譜法(2010年版藥典三部附錄Ⅲ B)測定。
10.1 色譜條件
用十八烷基硅烷鍵合硅膠填充劑(SG120,S-5μm,直徑4.6mm,長250mm);以70%乙腈溶液爲流動相;流速爲每分鐘1.2ml;檢測波長爲360nm;記錄時間爲30分鐘。
10.2 測定法
取戊二醛對照品適量,精密稱定.加水溶解並定量稀釋成每1ml中約含10μg的溶液;精密量取該溶液0.2ml、0.4ml、0.6ml、0.8ml、1.0ml.分別置試管中,各加水至1.0ml,精密加流動相1ml與2,4-二硝基苯肼溶液(稱取2,4-二硝基苯肼2.4g,加30%高氯酸溶液,溶解成100ml)0.1ml,立即於混合器上混勻,用0.45μm膜濾過。另取供試品適量,以每分鐘3000轉離心10分鐘,精密量取上清液1ml,自“精密加流動相1ml”起,同法操作。分別精密量取對照品溶液與供試品溶液各10μl,注入液相色譜儀,記錄色譜圖。
以戊二醛對照品溶液的濃度對其相應的峯面積作直線迴歸,求得直線迴歸方程,計算出供試品溶液中戊二醛含量。
10.3 【附註】
(1)配製戊二醛對照品溶液用的戊二醛用量0.1g(系經色譜純度測定後折算其含量爲100%)。
(2)直線迴歸相關係數應不低於0.99。
11 附錄Ⅵ J 磷酸三丁酯殘留量測定法
照氣相色譜法(2010年版藥典三部附錄Ⅲ C)測定。
11.1 色譜條件與系統適用性試驗
用酸改性聚乙二醇(20M)毛細管柱,1柱溫140℃,氣化室溫度190℃,火焰離子化檢測器或氮磷檢測器,檢測器溫度210℃,載氣(氮氣)流速爲每分鐘60ml,或根據儀器選擇檢測條件。理論板數按磷酸三丁酯峯計算應不低於5000,磷酸三丁酯蜂與磷酸三丙酯峯的分離瘦應不小於1.5,磷酸三丁酯對照品溶液連續進樣5次,所得磷酸三丁酯峯與磷酸三丙酯峯面積之比的相對標準偏差(RSD)應不大於5%。內標溶液的製備 取磷酸三丙酯適量,用正已烷溶解並定量稀釋製成每1ml中約含400μg的溶液。
11.2 測定法
精密量取供試品3ml,置具塞玻璃離心管中,精密加內標溶液50μl與1.5nmol/L高氯酸溶液0.75ml,振盪1分鐘;置37℃水浴保溫10分鐘後,再加正己烷4ml,振盪2分鐘;以每分鐘2000轉離心20分鐘,小心吸取上層正己烷,用空氣流將其濃縮至約0.2ml(不能加熱),取0.1μl注入氣相色譜儀。另取磷酸三丁酯對照品適量,精密稱定,加正已烷溶解並定量稀釋製成每1ml中約含600μg的溶液;精密量取該溶液10μl、20μl、40μl、60μl、80μl,分別置已精密加水3ml的具塞玻璃離心管中,再向各對照品管精密加內標溶液50μl,自“振盪1分鐘”起,同法操作。以各磷酸三丁酯對照品峯面積與內標峯面積比,對磷酸三丁酯對照品溶液濃度作直線迴歸,求得直線迴歸方程,計算出供試品中磷酸三丁酯含量(μg/ml)。
11.3 【附註】
(1)對照品溶液與供試品溶液的溶劑揮發的速度應儘量保持一致;若離心後,乳化仍未完全破除,可在振盪器上稍微振搖一下,再離心1次。
(2)直線迴歸相關係數應不低於0.99。
12 附錄Ⅵ K 辛酸鈉測定法
本法系用氣相色譜法測定供試品中辛酸鈉含量。照氣相色譜法(2010年版藥典三部附錄Ⅲ C)測定。
12.1 色譜條件與系統適用性試驗
用酸改性聚乙二醇(20M)毛細管柱,柱溫160℃,火焰離子化檢測器,檢測器溫度230℃,氣化室溫度230℃,載氣(氮氣)流速爲每分鐘35ml。辛酸峯與庚酸峯的分離度應大於1.5,辛酸峯的拖尾因子應爲0.95~1.20,辛酸對照品溶液連續進樣5次,所得辛酸峯與庚酸峯面積之比的相對標準偏差(RSD)應不大於5%。
12.2 內標溶液的製備
12.3 測定法
取供試品,用水準確稀釋成每升含蛋白質40~50g的溶液,即爲供試品溶液。精密量取供試品溶液0.5ml,加內標溶液30μl與1.5mol/L高氯酸溶液0.2ml,于振蕩器上混合1分鐘,加三氯甲烷4ml,加蓋,于振蕩器上劇烈混合2分鐘,以每分鐘3000轉離心20分鐘,除去上層水相,小心將三氯甲烷層傾入10ml試管中,將三氯甲烷揮發至幹,加三氯甲烷100μl溶解殘渣,取0.1μl注入氣相色譜儀。另取辛酸對照品約0.15g,精密稱定,置10ml量瓶中,用三氯甲烷溶解並稀釋至刻度,即爲辛酸對照品溶液。精密量取辛酸對照品溶液10μl、20μl、30μl、40μl、50μl,各精密加內標溶液30μl,于振蕩器上混合1分鐘,加三氯甲烷4ml,將三氯甲烷揮發至幹,各加三氯甲烷100μl溶解殘渣,同法操作。
以各辛酸對照品溶液峯面積與內標峯面積比對各辛酸對照品溶液辛酸量(μg)作直線迴歸,求得直線迴歸方程,計算出供試品溶液辛酸絕對量(A),再按下式計算供試品辛酸鈉含量:

B爲取樣量,即爲0.5ml;
n爲供試品稀釋倍數;
c爲供試品蛋白質含量,g/ml;
12.4 【附註】
(1)1mmol辛酸相當於1mmol辛酸鈉。
(2)對照品溶液與供試品溶液的溶劑揮發的速度應儘量保持一致。
(3)直線迴歸相關係數應不低於0.99。
13 附錄Ⅵ L 遊離甲醛測定法
本法系依據品紅亞硫酸在酸性溶液中能與甲醛生成紫色復合物,用比色法測定供試品中游離甲醛含量。
13.1 對照品溶液的製備
精密量取已標定的甲醛溶液適量,置500ml量瓶中,用水稀釋至刻度,搖勻,製成0.05%甲醛對照品貯備液。
臨用前,精密量取甲醛對照品貯備液10ml,置100ml量瓶中,加水稀釋至刻度,搖勻,作爲甲醛對照品溶液。測定法 精密量取供試品1ml,用水稀釋至甲醛含量約爲0.005%,即爲供試品溶液。精密量取供試品溶液1ml,置50ml具塞試管中,加水4ml,加品紅亞硫酸溶液10ml,混合酸溶液(量取水783ml,置燒杯內,緩緩注入鹽酸42ml、硫酸175ml,混勻)10ml,搖勻,於25℃放置3小時,照紫外-可見分光光度法(2010年版藥典三部附錄Ⅱ A),在波長590nm處測定吸光度。
精密量取0.005%甲醛對照品溶液0.5ml、1.0ml、1.5ml、2.0ml,分別置。50ml具塞試管中,加水至5ml,自“加品紅亞硫酸溶液10ml”起,同法操作。
以甲醛對照品溶液的濃度對相應的吸光度作直線迴歸,將供試品溶液的吸光度代入直線迴歸方程,計算供試品中的遊離甲醛含量。
【附註】
(1)品紅亞硫酸溶液的製備及二氧化硫含量的標定 稱取鹼性品紅4.5g,於3000ml錐形瓶中,加水1500ml,振搖或加溫使品紅全部溶解,待冷後,加亞硫酸鈉10g,搖勻,靜置5~10分鐘,再加入3nmol/L硫酸溶液40ml,搖勻,以橡皮塞塞緊瓶口,放置過夜,如有顏色,加骨炭5~10g迅速搖勻,以布氏漏斗快速抽濾,即得品紅亞硫酸溶液。品紅亞硫酸溶液中的SO2含量可控制在28~48mmol/L(SO2含量過多可通空氣驅除,過少可通人SO2)。
二氧化硫(SO2)含量測定 量取品紅亞硫酸溶液10ml於錐形瓶內,加水20ml,澱粉指示液5ml,用碘滴定液(0.05mol/L)滴定至呈淺藍色,按下式計算SO2的含量;
SO2的含量(mmol/L)=50×V×c
式中 V爲消耗碘滴定液(0.05mol/L)的體積,ml;
c爲碘滴定液的濃度,mol/L。
(2)甲醛溶液的標定 取甲醛溶液約1.5ml,精密稱定,置錐形瓶中,加水10ml、過氧化氫溶液25ml與溴麝香草酚藍指示液2滴,滴加氫氧化鈉滴定液(,1mol/L)至溶液顯藍色;再精密加入氫氧化鈉滴定液(1mol/L)25ml,瓶口置一玻璃小漏斗,置水浴中加熱15分鐘,不時振搖,冷卻,用水洗滌漏斗,加溴麝香草酚藍指示液2滴,用鹽酸滴定液(1mol/L)滴定至溶液顯黃色,並將滴定的結果用空白試驗校正。每1ml氫氧化鈉滴定液(1mol/L)相當於30.03mg的甲醛。
(3)對照品溶液和供試品溶液與品紅亞硫酸溶液的顯色時間有時不一致,測定時,顯色慢者應酌情早加品紅亞硫酸溶液。
(4)供試品中如含有酚紅,標準管中應予以校正。附錄Ⅵ M 苯酚測定法本法系依據溴酸鹽溶液與鹽酸反應產生溴,遇苯酚生成三溴苯酚,過量的溴與碘化鉀反應釋出碘,析出的碘用硫代硫酸鈉滴定液滴定,根據硫代硫酸鈉滴定液的消耗量,可計算出供試品中苯酚的含量。
13.2 測定法
精密量取供試品1ml,置具塞錐形瓶中,加水50ml,精密加入0.02mol/L溴溶液(稱取溴酸鉀0.56g,加溴化鉀3g,加水溶解並稀釋至1000ml)15~25ml(供試品含苯酚量0.3%~0.5%時加25ml,小於0.3%則加15ml),沿瓶壁加入6mol/L鹽酸溶液10ml,搖勻,密塞,在暗處放置30分鐘後,加25%碘化鉀溶液2ml於具塞錐形瓶頸口,稍啓瓶塞,使流下,密塞,搖勻。以少量水洗瓶頸,用硫代硫酸鈉滴定液(0.02mol/L)滴定至近終點時,加澱粉指示液約0.5ml,滴定至藍色消失,並將滴定的結果用空白試驗校正。
按下式計算:

【附註】
(1)硫代硫酸鈉滴定液(0.1mol/L)的製備及標定 稱取硫代硫酸鈉26g與無水碳酸鈉0.20g,加新沸過的冷水適量溶解並稀釋至1000ml,搖勻,放置1個月後濾過。取在120℃乾燥至恆重的基準重鉻酸鉀0.15g,精密稱定,置碘瓶中,加水50ml溶解,加碘化鉀2.0g,輕輕振搖使溶解,加稀硫酸(5.7→100) 40ml,搖勻,密塞;在暗處放置10分鐘後,加水250ml稀釋,用本液滴定至近終點時,加澱粉指示液(稱取可溶性澱粉0.5g,加水5ml混懸後緩緩傾入100ml沸水中,隨加隨攪拌,繼續煮沸2分鐘,冷卻,傾取上層清液。本液應臨用配製)3ml,繼續滴定至藍色消失而顯亮綠色,並將滴定的結果用空白試驗校正。每1ml硫代硫酸鈉滴定液(0.1mol/L)相當於4.903mg重鉻酸鉀。根據本液的消耗量與重鉻酸鉀的取用量,算出本液的濃度,即得。
(2)硫代硫酸鈉滴定液(0.02mol/L)的製備 精密量取硫代硫酸鈉滴定液(0.1mol/L)100ml,加水準確稀釋至500ml,搖勻。
(3)可做限度測定。
14 附錄Ⅵ N 間甲酚測定法
本法系依據4-氨基安替比林、鐵氰化鉀在鹼性條件下與間甲酚反應生成一種紅色物質,用比色法測定供試品中間甲酚含量。
14.1 測定法
精密量取一定體積的供試品,置試管中,定量稀釋50倍,即爲供試品溶液。量取供試品溶液1.0ml,加水5.0ml,混勻,依次加pH9.8緩衝液(稱取無水碳酸鈉6.36g、碳酸氫鈉3.36g,加水溶解並稀釋至800ml,用1mol/L鹽酸調pH值至9.8後,再加水至1000ml)、0.3% 4-氨基安替比林溶液、1.2%鐵氰化鉀溶液及1mol/L磷酸二氫鉀溶液各1.0ml,混勻,於室溫避光放置10分鐘,照紫外-可見分光光度法(2010年版藥典三部附錄Ⅱ A),在波長510nm處測定吸光度。
精密量取間甲酚對照品溶液(取間甲酚適量,精密稱定,置量瓶中,加水溶解並稀釋至每1ml含10μg)1.0ml、2.0ml、3.0ml、4.0ml、5.0ml、6.0ml,分別置試管中,加水補至6.0ml,自“依次加pH9.8緩衝液”起,同法操作,測定各管的吸光度。
以間甲酚對照品溶液的系列濃度對其相應的吸光度作直線迴歸,將供試品溶液的吸光度代入直線迴歸方程,得供試品的間甲酚含量(mg/ml)。
15 附錄Ⅵ P 人血液製品中糖及糖醇測定法
照離子色譜法(2010年版藥典三部附錄Ⅲ E)測定。
15.1 色譜條件與系統適用性試驗
用苯乙烯-二乙烯基苯共聚物爲基質的陽離子交換色譜柱(H+),粒度9μm或8μm,內徑7.8mm,柱長300mm;柱溫50℃(測定蔗糖含量時,柱溫爲20~30℃);流動相爲0.004mol/L硫酸溶液,流速爲每分鐘0.8ml;示差折光檢測器。取2%麥芽糖1ml和1.5%磺基水楊酸1ml的混合物20μl,注入色譜柱,記錄色譜圖,麥芽糖與磺基水楊酸兩峯間的分離度應大於1.5,拖尾因子按麥芽糖峯計算應爲0.95~1.50。
15.2 對照品溶液的製備
(1)麥芽糖對照品溶液 分別取經減壓乾燥至恆重的麥芽糖對照品1.0g、2.0g、3.0g,精密稱定,各置100ml量瓶中,分別加水溶解並稀釋至刻度,搖勻,即得。
(2)葡萄糖對照品溶液 分別取經減壓乾燥至恆重的葡萄糖對照品0.5g、1.0g、1.5g,精密稱定,各置100ml量瓶中,分別加水溶解並稀釋至刻度,搖勻,即得。
(3)山梨醇對照品溶液 分別取經減壓乾燥至恆重的山梨醇對照品0.5g、1.0g、1.5g,精密稱定,各置100ml量瓶中,分別加水溶解並稀釋至刻度,搖勻,即得。
(4)蔗糖對照品溶液 分別取經減壓乾燥至恆重的蔗糖對照品1.0g、2.0g、3.0g,精密稱定,各置100ml量瓶中,分別加水溶解並稀釋至刻度,搖勻,即得。
15.3 供試品溶液的製備
精密量取供試品1ml,加1.5%磺基水楊酸4.0ml,混勻,室溫放置至少2小時,以每分鐘3000轉離心10分鐘,取上清液,即得。
15.4 測定法
精密量取對照品溶液與供試品溶液,分別注入液相色譜儀,記錄色譜圖;進樣量爲20μl。
以各對照品溶液濃度(g/L)對其相應的的峯面積作直線迴歸,求得直線迴歸方程,計算出供試品溶液中糖或糖醇含量(A),再按下列公式計算:
供試品糖或糖醇含量(g/L)=A×n
n爲供試品稀釋倍數。
【附註】
(1)根據供試品的糖含量,對照品和供試品的取量可做適當調整。
(2)直線迴歸相關係數應不低於0.999。
(3)不同廠家的陽離子交換色譜柱(H+)的流速、流動相、柱溫等會有所不同,可根據色譜柱介紹對色譜條件進行適當調整。
16 附錄Ⅵ Q 人血白蛋白多聚體測定法
照分子排阻色譜法(2010年版藥典三部附錄Ⅲ D)測定。
16.1 色譜條件與系統適用性試驗
用親水硅膠高效體積排阻色譜柱(SEC,排阻極限300kD,粒度10μm),柱直徑7.5mm,長60cm;以含1%異丙醇的pH7.0、0.2mol/L磷酸鹽緩衝液(量取0.5mol/L磷酸二氫鈉200ml、0.5mol/L磷酸氫二鈉420ml、異丙醇15.5ml及水914.5ml,混勻]爲流動相;檢測波長爲280nm;流速爲每分鐘0.6ml。取每1ml含蛋白質12mg的人血白蛋白溶液20μl.注入色譜柱,記錄色譜圖,人血白蛋白單體峯與二聚體峯間的分離度應大於1.5,拖尾因子按人血白蛋白單體峯計算應爲0.95~1.40。
16.2 測定法
17 附錄Ⅵ R 人免疫球蛋白類製品IgG單體加二聚體測定法
本法系用分子排阻色譜法測定人免疫球蛋白類製品IgG單體加二聚體含量。
照分子排阻色譜法(2010年版藥典三部附錄Ⅲ D)測定。
17.1 色譜條件與系統適用性試驗
用親水硅膠高效體積排阻色譜柱(SEC,排阻極限300kD,粒度10μm),柱直徑7.5mm,長60cm。以含1%異丙醇的pH7.0、0.2mol/L磷酸鹽緩衝液(量取0.5mol/L磷酸二氫鈉200ml、0.5mol/L磷酸氫二鈉420ml、異丙醇15.5ml及水914.5ml,混勻]爲流動相,檢測波長爲280nm,流速爲每分鐘0.6ml。分別取每1ml含蛋白質爲12mg的人免疫球蛋白、人血白蛋白溶液各20μl,分別注入色譜柱,記錄色譜圖。人免疫球蛋白對照品單體峯與裂解體峯的分離度應大於1.5,人血白蛋白對照品單體峯與二聚體峯的分離度應大於1.5,拖尾因子按人血白蛋白單體峯計算應爲0.95~1.40。
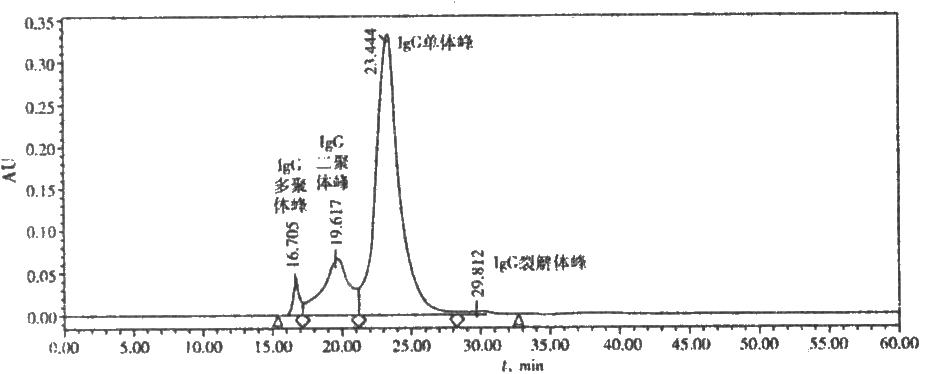
圖 人免疫球蛋白IgG標準圖譜
17.2 測定法
取供試品適量,用流動相稀釋成每1ml約含蛋白質12mg的溶液,取20μl,注入色譜柱,記錄色譜圖60分鐘。按面積歸一法計算,色譜圖中單體加二聚體峯的含量,即爲IgG單體加二聚體含量。圖譜各蜂的界限爲兩峯間最低點到基線的垂直線。主峯爲IgG單體;相對保留時間約0.85的峯爲二聚體。
18 附錄Ⅵ S 人免疫球蛋白中甘氨酸含量測定法
本法系依據過量的6-氨基喹啉基-N-羥基琥珀酰亞氨基氨基甲酸酯(AQC)在一定條件下和氨基酸形成穩定的衍生產物(柱前衍生),用高效液相色譜法測定衍生產物,根據衍生產物的含量計算人免疫球蛋白中甘氨酸含量。
照高效液相色譜法(2010年版藥典三部附錄Ⅲ B)測定。
18.1 色譜條件與系統適應性試驗
用十八烷基硅烷鍵合硅膠爲基質的C18反相色譜柱,粒度4μm,內徑3.9mm,柱長150mm;柱溫爲37℃;以140mmol/L醋酸鈉、17mmol/L三乙胺(pH 5.65)、1μg/ml乙二胺四乙酸二鈉爲流動相A液,以100%乙腈爲流動相B液,以純水爲流動相C液,流速爲每分鐘1.0ml,梯度洗脫32分鐘(梯度表),檢測波長爲248nm。甘氨酸與相鄰色譜峯之間分離度應大於1.5;拖尾因子(T)爲0.95~1.40(甘氨酸和α-氨基丁酸峯);RSD應不大於2.0%(甘氨酸對照品峯面積測量值)。
18.2 內標溶液的製備
精密稱取α-氨基丁酸對照品0.4g[1],加超純水定容至100ml。
18.3 對照品溶液的製備
(1)精密稱取甘氨酸對照品2.5g,加超純水定容至100ml。
(2)精密量取(1)項溶液1.0ml,加9.0ml 1.5%磺基水楊酸,混勻靜置2小時以上,以每分鐘3000轉離心10分鐘,留取上清液備用。
(3)精密量取(2)項上清液0.4ml、0.8ml、1.0ml、1.2ml、1.6ml,分別置10ml量瓶中,用純水定容。
(4)精密量取(3)項溶液各0.1ml,加純水0.4ml,內標溶液0.02ml,混勻備用。
(5)精密量取(4)項溶液10μl放入衍生管中,加硼酸緩衝液(pH8~10)70μl渦旋混合,並加入20μl AQC衍生劑渦旋混合15秒,即爲對照品溶液。
18.4 供試晶溶液的製備
(1)精密量取供試品溶液1.0ml,加1.5%磺基水楊酸9.0ml,混勻靜置2小時以上,以每分鐘3000轉離心10分鐘,留取上清液備用。
(2)精密量取(1)項上清液1.0ml,置10ml量瓶中,用純水定容。
(3)精密量取(2)項溶液0.1ml,加0.4ml純水,加內標溶液0.02ml,混勻後,精密量取10μl放入衍生管中加70μl硼酸緩衝液渦旋混合並加入20μl AQC衍生劑渦旋混合15秒,即爲供試品溶液。
18.5 測定法
精密量取對照品溶液與供試品溶液,分別注入液相色譜儀,記錄色譜圖32分鐘。進樣量爲10μl。按內標法計算。
梯度表
時間/分鐘 | 流速/ml·min-1 | A液/% | B液/% | C液/% | 曲線 |
起始 | 1.0 | 100 | 0 | 0 | |
0.5 | 1.0 | 99.0 | 1.0 | 0 | 瞬時 |
18.00 | 1.0 | 95.0 | 5.0 | 0 | |
19.00 | 1.0 | 91.0 | 9.0 | 0 | |
22.00 | 1.0 | 83.0 | 17.0 | 0 | |
25.00 | 1.0 | 0 | 60.0 | 40.0 | 瞬時 |
28.00 | 1.0 | 100 | 0 | 0 | 瞬時 |
32.00 | 1.0 | 100 | 0 | 0 |
【附註】
(1)甘氨酸含量測定應採用柱前衍生及內標法,除本法要求外,衍生劑也可選用異硫氰酸苯酯、鄰苯二甲醛;內標物也可選用正纈氨酸;C18反相色譜柱的粒度也可選用5μm或亞二微米。根據液相色譜系統、C18反相色譜柱規格、衍生劑及內標物的不同可以調整相應的色譜條件。
(2)直線迴歸相關係數應不低於0.999。
19 附錄Ⅵ T 對羥基苯甲酸甲酯、對羥基苯甲酸丙酯含量測定法
本法系用氣相色譜法測定供試品中對羥基苯甲酸甲酯及對羥基苯甲酸丙酯含量。
照氣相色譜法(2010年版藥典三部附錄Ⅲ C)測定。
19.1 色譜條件與系統適用性試驗
用塗布100%聚二甲基硅氧烷石英毛細管柱,柱溫180℃,氣化室溫度250℃;氫離子化火焰檢測器,檢測器溫度300℃。載氣爲氮氣,流速爲每分鐘20ml。進樣方式採用分流進樣,進樣量爲1μl。內標物(對苯二酚)與對羥基苯甲酸甲酯及對羥基苯甲酸丙酯之間的分離度均應大於1.5,對羥基苯甲酸甲酯及對羥基苯甲酸丙酯的對照品溶液連續進樣5次,所得對羥基苯甲酸甲酯及對羥基苯甲酸丙酯峯面積與對苯二酚峯面積之比的相對標準偏差應不大於5%。
19.2 內標溶液的製備
取對苯二酚50mg,精密稱定,用無水乙醇定容至50ml,製成每1ml約含有1mg的內標溶液。
19.3 對照品溶液的製備
取對羥基苯甲酸甲酯0.1g、對羥基苯甲酸丙酯0.01g,精密稱定,用無水乙醇定容至10ml,即得約1.00%對羥基苯甲酸甲酯的溶液、約0.10%對羥基苯甲酸丙酯的溶液。
19.4 校正因子測定用對照溶液的製備
取對照品溶液60μl,內標溶液100μl,加純化水840μl,即得含內標物100μg/ml、對羥基苯甲酸甲酯約0.06%、對羥基苯甲酸丙酯約0.006%的校正因子測定用對照溶液。
19.5 測定法
取供試品840μl,加入內標溶液100μl、無水乙醇60μl,混勻,取1μl注入氣相色譜儀,另取1μl校正因子測定用對照溶液,同法操作,按內標加校正因子測定法計算對羥基苯甲酸甲酯及對羥基苯甲酸丙酯含量。
20 附錄Ⅵ U 重組人粒細胞刺激因子蛋白質含量測定法
本法採用高效液相色譜法測定供試品中重組人粒細胞刺激因子蛋白質含量。
照高效液相色譜法(2010年版藥典三部附錄Ⅲ B)測定。
20.1 色譜條件
色譜柱採用十八烷基硅烷鍵合硅膠爲填充劑,孔徑30nm,粒度5μm,直徑4.6mm,長250mm;柱溫爲30℃±5℃,供試品保存溫度爲2~8℃;以0.1%三氟乙酸的水溶液爲流動相A液,以0.1%三氟乙酸的乙腈溶液爲流動相B液;流速爲1ml/min;檢測波長214nm;按下表進行梯度洗脫。
編號 | 時間/分鐘 | A/% | B/% |
1 | 0 | 60 | 40 |
2 | 40 | 20 | 80 |
3 | 45 | 0 | 100 |
4 | 50 | 60 | 40 |
5 | 60 | 60 | 40 |
20.2 檢查法
取1支標準品,按介紹復溶。用20mmol/L的醋酸-醋酸鈉緩衝液(pH4.0)將標準品及供試品調節至相同蛋白質濃度,將供試品溶液與標準品溶液以相同
體積分別注入液相色譜儀(進樣體積不小於10μl,進樣量4~6μg),按上表進行梯度洗脫。標準品溶液、供試品溶液均進樣3次,記錄色譜圖並計算峯面積。按下式計算重組人粒細胞刺激因子蛋白質含量(μg/ml):

nX爲供試品溶液的稀釋倍數。
21 附錄Ⅵ V 殘留溶劑測定法
藥品中的殘留溶劑係指在原料藥或輔料的生產中,以及在製劑製備過程中使用的,但在工藝過程中未能完全去除的有機溶劑。藥品中常見的殘留溶劑及限度見附表1,除另有規定外,第一、第二、第三類溶劑的殘留限度應符合附表1中的規定;對其他溶劑,應根據生產工藝的特點,制定相應的限度,使其符合產品規範、藥品生產質量管理規範(GMP)或其他基本的質量要求。
本法照氣相色譜法(2010年版藥典三部附錄Ⅲ C)測定。
21.1 色譜柱
21.1.1 1.毛細管柱
除另有規定外,極性相近的同類色譜柱之間可以互換使用。
(1)非極性色譜柱 固定液爲100%的二甲基聚硅氧烷的毛細管柱。
(2)極性色譜柱 固定液爲聚乙二醇(PEG-20M)的毛細管柱。
(3)中極性色譜柱 固定液爲(35%)二苯基-(65%)甲基聚硅氧烷、(50%)二苯基-(50%)二甲基聚硅氧烷、(35%)二苯基-(65%)二甲基聚硅氧烷、(14%)氰丙基苯基-(86%)二甲基聚硅氧烷、(6%)氰丙基苯基-(94%)二甲基聚硅氧烷的毛細管柱等。
(4)弱極性色譜 柱固定液爲(5%)苯基-(95%)甲基聚硅氧烷、(5%)二苯基-(95%)二甲基硅氧烷共聚物的毛細管柱等。
21.1.2 2.填充柱
以直徑爲0.18~0.25mm的二乙烯苯-乙基乙烯苯型高分子多孔小球或其他適宜的填料作爲固定相。
21.2 系統適用性試驗
(1)用待測物的色譜峯計算,毛細管色譜柱的理論板數一般不低於5000;填充柱的理論板數一般不低於1000。
(2)色譜圖中,待測物色譜峯與其相鄰色譜峯韻分離度應大於1.5。
(3)以內標法測定時,對照品溶液連續進樣5次,所得待測物與內標物峯面積之比的相對標準偏差(RSD)應不大於5%;若以外標法測定,所得待測物峯面積的RSD應不大於10%。
21.3 供試品溶液的製備
21.3.1 1.頂空進樣
除另有規定外,精密稱取供試品0.1~1g,通常以水爲溶劑;對於非水溶性藥物,可採用N,N-二甲基甲酰胺、二甲基亞碸或其他適宜溶劑;根據供試品和待測溶劑的溶解度,選擇適宜的溶劑且應不干擾待測溶劑的測定。根據各品種項下殘留溶劑的限度規定配製供試品溶液,其濃度應滿足系統定量測定的需要。
21.3.2 2.溶液直接進樣
精密稱取供試品適量,用水或合適的有機溶劑使溶解;根據各品種項下殘留溶劑的限度規定配製供試品溶液,其濃度應滿足系統定量測定的需要。
21.4 對照品溶液的製備
精密稱取各品種項下規定檢查的有機溶劑適量,採用與製備供試品溶液相同的方法和溶劑製備對照品溶液;如用水作溶劑,應先將待測有機溶劑溶解在50%二甲基亞碸或N,N-二甲基甲酰胺溶液中,再用水逐步稀釋。若爲限度檢查,根據殘留溶劑的限度規定確定對照品溶液的濃度;若爲定量測定,爲保證定量結果的準確性,應根據供試品中殘留溶劑的實際殘留量確定對照品溶液的濃度;通常對照品溶液色譜蜂面積不宜超過供試品溶液中對應的殘留溶劑色譜蜂面積的2倍。必要時,應重新調整供試品溶液或對照品溶液的濃度。
21.5 測定法
21.5.1 第一法(毛細管柱頂空進樣等溫法)
當需要檢查有機溶劑的數量不多,且極性差異較小時,可採用此法。
色譜條件 柱溫一般爲40~100℃;常以氮氣爲載氣,流速爲每分鐘1.0~2.0ml;以水爲溶劑時頂空瓶平衡溫度爲70~85℃,頂空瓶平衡時間爲30~60分鐘;進樣口溫度爲200℃;如採用火焰離子化檢測器(FID),溫度爲250℃。
測定法 取對照品溶液和供試品溶液,分別連續進樣不少於2次,測定待測峯的峯面積。
對色譜圖中未知有機溶劑的鑑別,可參考附表2進行初篩。
21.5.2 第二法(毛細管柱頂空進樣系統程序升溫法)
當需要檢查的有機溶劑數量較多,且極性差異較大時,可採用此法。
色譜條件 柱溫一般先在40℃維持8分鐘,再以每分鐘8℃的升溫速率升至120℃,維持10分鐘;以氮氣爲載氣,流速爲每分鐘2.0ml;以水爲溶劑時頂空瓶平衡溫度爲70~85℃,頂空瓶平衡時間爲30~60分鐘;進樣口溫度爲200℃;,如採用FID檢測器,進樣口溫度爲250℃。具體到某個品種的殘留溶劑檢查時,可根據該品種項下殘留溶劑的組成調整升溫程序。
測定法 取對照品溶液和供試品溶液,分別連續進樣不少於2次,測定待測峯的峯面積。
對色譜圖中未知有機溶劑的鑑別,可參考附表3進行初篩。
21.5.3 第三法(溶液直接進樣法)
可採用填充柱,亦可採用適宜極性的毛細管柱。
測定法 取對照品溶液和供試品溶液,分別連續進樣2~3次,測定待測峯的峯面積。
21.6 計算法
(1)限度檢查 除另有規定外;按各品種項下規定的供試品溶液濃度測定。以內標法測定時,供試品溶液所得被測溶劑峯面積與內標峯面積之比不得大於對照品溶液的相應比值。以外標法測定時,供試品溶液所得被測溶劑峯面積不得大於對照品溶液的相應峯面積。
(2)定量測定 按內標法或外標法計算各殘留溶劑的量。
21.7 【附註】
(1)除另有規定外,頂空條件的選擇:
①應根據供試品中殘留溶劑的沸點選擇頂空平衡溫度。對沸點較高的殘留溶劑,通常選擇較高的平衡溫度;但此時應兼顧供試品的熱分解特性,儘量避免供試品產生的揮發性熱分解產物對測定的干擾。
②頂空平衡時間一般爲30~45分鐘,以保證供試品溶液的氣一液兩相有足夠的時間達到平衡。頂空平衡時間通常不宜過長,如超過60分鐘,可能引起頂空瓶的氣密性變差,導致定量準確性的降低。
(2)定量方法的驗證 當採用頂空進樣時,供試品與對照品處於不完全相同的基質中,故應考慮氣液平衡過程中的基質效應(供試品溶液與對照品溶液組成差異對頂空氣-液平衡的影響)。由於標準加入法可以消除供試品溶液基質與對照品溶液基質不同所致的基質效應的影響,故通常採用標準加入法驗證定量方法的準確性;當標準加入法與其他定量方法的結果不一致時,應以標準加入法的結果爲準。
(3)干擾峯的排除 供試品中的未知雜質或其揮發性熱降解物易對殘留溶劑的測定產生干擾。干擾作用包括在測定的色譜系統中未知雜質或其揮發性熱降解物與待測物的保留值相同(共出峯);或熱降解產物與待測物的結構相同(如甲氧基熱裂解產生甲醇)。當測定的殘留溶劑超出限度,但未能確定供試品中是否有未知雜質或其揮發性熱降解物對測定有干擾作用時,應通過試驗排除干擾作用的存在。對第一類干擾作用,通常採用在另一種極性不同的色譜柱系統中對相同供試品再進行測定,比較不同色譜系統中測定結果的方法。如兩者結果一致,則可以排除測定中有共出峯的干擾;如兩者結果不一致,則表明測定中有共出峯的干擾。對第二類干擾作用,通常要通過測定已知不含該溶劑的對照樣品來加以判斷。
(4)含氮鹼性化合物的測定 普通氣相色譜儀中的不鏽鋼管路、進樣器的襯管等對有機胺等含氮鹼性化合物具有較強的吸附作用,致使其檢出靈敏度降低,應採用惰性的硅鋼材料或鎳鋼材料管路;採用溶液直接進樣法測定時,供試品溶液應不呈酸性,以免待測物與酸反應後不易汽化。
通常採用弱極性的色譜柱或其填料預先經鹼處理過的色譜柱分析含氮鹼性化合物,如果採用胺分析專用柱進行分析,效果更好。
對不宜採用氣相色譜法測定的含氮鹼性化合物,如N-甲基吡咯烷酮等,可採用其他方法如離子色譜法等測定。
(5)檢測器的選擇 對含鹵素元素的殘留溶劑如三氯甲烷等,採用電子捕獲檢測器(ECD),易得到高的靈敏度。
(6)由於不同的實驗室在測定同一供試品時可能採用了不同的實驗方法,當測定結果處於合格與不合格邊緣時,以採用內標法或標準加入法爲準。
(7)頂空平衡溫度一般應低於溶解供試品所用溶劑的沸點10℃以下,能滿足檢測靈敏度即可;對於沸點過高的溶劑,如甲酰胺、2-甲氧基乙醇、2-乙氧基乙醇、乙二醇、N-甲基吡咯烷酮等,用頂空進樣測定的靈敏度不如直接進樣,一般不宜用頂空進樣方式測定。
(8)利用保留值定性是氣相色譜中最常用的定性方法。色譜系統中載氣的流速、載氣的溫度和柱溫等的變化都會使保留值改變,從而影響定性結果。校正相對保留時間(RART)只受柱溫和固定相性質的影響,以此作爲定性分析參數較可靠。應用中通常選用甲烷測定色譜系統的死體積(tO):
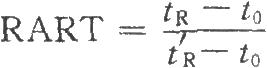
式中tR爲組分的保留時間;
tR'爲參比物的保留時間。
22 附表1 藥品中常見的殘留溶劑及限度
溶劑名稱 | 限度/% |
第一類溶劑(應該避免使用) | |
苯 | 0.0002 |
四氯化碳 | 0.0004 |
0.0005 | |
0.0008 | |
0.15 | |
第二類溶劑(應該限制使用) | |
0.041 | |
0.036 | |
0.006 | |
0.388 | |
0.187 | |
0.06 | |
0.01 | |
N,N-二甲基乙酰胺 | 0.109 |
0.088 | |
二氧六環 | 0.038 |
溶劑名稱 | 限度/% |
第二類溶劑(應該限制使用) | |
2-乙氧基乙醇 | 0.016 |
0.062 | |
甲酰胺 | 0.022 |
0.029 | |
0.3 | |
2-甲氧基乙醇 | 0.005 |
甲基丁基酮 | 0.005 |
0.118 | |
0.053 | |
0.005 | |
0.02 | |
0.016 | |
四氫化萘 | 0.01 |
四氫呋哺 | 0.072 |
甲苯 | 0.089 |
1,1,2-三氯乙烯 | 0.008 |
二甲苯① | 0.217 |
溶劑名稱 | 限度/% |
第三類溶劑(藥品GMP或其他質量要求限制使用) | |
0.5 | |
0.5 | |
甲氧基苯 | 0.5 |
0.5 | |
仲丁醇 | 0.5 |
乙酸丁酯 | 0.5 |
叔丁基甲基醚 | 0.5 |
異丙基苯 | 0.5 |
0.5 | |
0.5 | |
0.5 | |
0.5 | |
0.5 | |
0.5 | |
0.5 | |
0.5 | |
0.5 | |
0.5 | |
0.5 |
溶劑名稱 | 限度/% |
第三類溶劑(藥品GMP或其他質量要求限制使用) | |
丁酮 | 0.5 |
甲基異丁基酮 | 0.5 |
異丁醇 | 0.5 |
0.5 | |
正戊醇 | 0.5 |
正丙醇 | 0.5 |
0.5 | |
乙酸丙酯 | 0.5 |
第四類溶劑(尚無足 夠毒理學資料)② | |
1,1-二乙氧基丙烷 | |
1,1-二甲氧基甲烷 | |
異辛烷 | |
甲基異丙基酮 | |
甲基四氫呋喃 | |
三氯醋酸 | |
三氟醋酸 |
①通常含有60%間二甲苯、14%對二甲苯、9%鄰二甲苯和17%乙苯。
②藥品生產企業在使用時應提供該類溶劑在製劑中殘留水平的合理性論證報告。
23 附表2 常見有機溶劑在等溫法測定時相對於丁酮的保留值參考值
非極性色譜柱 | ||
溶劑名稱 | tR/min | RART |
柱溫40℃ | ||
正丙醇 叔丁基甲基醚 丁酮 仲丁醇 異丁醇 甲基異丙基酮 苯 四氯化碳 甲基四氫呋喃 二氧六環 異辛烷 乙酸丙酯 甲基異丁基酮 甲苯 正戊醇 | 1.828 2.090 2.179 2.276 2.356 2.487 2.489 2.522 2.584 2.609 2.635 2.655 2.807 2.982 3.109 3.252 3.449 3.666 3.898 3.908 3.913 3.954 4.264 4.264 4.517 4.808 4.976 4.985 5.281 5.311 5.340 5.470 5.583 5.676 6.760 6.823 6.957 7.434 7.478 8.628 8.738 8.870 9.283 11.180 11.382 1.54 | 0.126 0.268 0.315 0.368 0.411 0.481 0.482 0.501 0.534 0.547 0.561 0.572 0.654 0.748 0.817 0.894 1.000 1.117 1.242 1.247 1.250 1.272 1.439 1.440 1.576 1.733 1.823 1.828 1.988 2.004 2.019 2.089 2.150 2.201 2.785 2.819 2.891 3.148 3.172 3.792 3.851 3.922 4.145 5.168 5.276 |
柱溫80℃ | ||
甲基丁基酮 乙酸丁酯 甲氧基苯 | 3.611 3.859 4.299 5.253 7.436 | 2.099 2.345 2.778 3.726 5.890 |
續表
極性色譜柱 | ||
溶劑名稱 | tR/min | RART |
柱溫40℃ | ||
異辛烷 叔丁基甲基醚 甲基環已烷 甲基四氫呋喃 四氯化碳 丁酮 甲基異丙基酮 苯 乙酸丙酯 甲基異丁基酮 仲丁醇 甲苯 正丙酵 二氧六環 乙酸丁酯 甲基丁基酮 | 1.682 1.787 1.842 1.926 1.943 2.005 2.021 2.159 2.209 2.243 2.405 2.876 2.967 3.000 3.347 3.403 3.481 3.635 3.6a3 3.810 3.980 4.062 4.079 4.604 4.716 4.758 4.822 4.975 4.977 6.020 6.643 7.202 7.368 7.497 7.985 8.390 8.746 9.238 10.335 10.827 11.012 11.486 1.602 | 0.032 0.075 0.097 0.131 0.138 0.163 0.169 0.225 0245 0.259 0.324 0.515 0.551 0.564 0.705 0.727 0.758 0.821 0.828 0.891 0.960 0.993 1.000 1.212 1.257 1.274 1.300 1.362 1.362 1.784 2.035 2.261 2.328 2.380 2.577 2.740 2.884 3.083 3.526 3.724 3.799 3.990 |
柱溫80℃ | ||
異丁醇 異丙基苯 正戊醇 | 3.577 4.460 4.885 5.288 5.625 5.934 6.439 7.332 | 3.045 4.334 4.948 5.543 6.035 6.486 7.223 8.527 |
續表
24 附表3 常見有機溶劑在程序升溫法測定時相對於丁酮的保留值參考值
非極性色譜柱 | ||
溶劑名稱 | tR/min | RART |
正丙醇 叔丁基甲基醚 丁酮 仲丁醇 異丁醇 甲基異丙基酮 苯 四氯化碳 甲基四氫呋喃 異辛烷 | 1.846 2.121 2.201 2.303 2.401 2.512 2.519 2.544 2.611 2.623 2.665 2.674 2.839 3.051 3.128 3.302 3.507 3.756 3.966 3.971. 3.981 4.005 4.387 4.397 4.6124 4.843 5.087 5.099 5.380 5.398 5.402 5.501 5.649 5.739 6.815 6.928 | 0.127 0.272 0.314 0.367 0.419 0.477 0.481 0.494 0.529 0.535 0.558 0.562 0.649 0.760 0.801 0.892 1.000 1.131 1.241 1.244 1.249 1.262 1.462 1.468 1.581 1.702 1.830 1.837 1.984 1.994 1.996 2.048 2.126 2.173 . 2.738 2.798 |
續表
非極性色譜柱 | ||
溶劑名稱 | tR/min | RART |
二氧六環 乙酸丙酯 甲基異丁基酮 甲苯 正戊醇 甲基丁基酮 乙酸丁酯 甲氧基苯 異丙基苯 四氫化萘 | 6.928 7.563 7.583 8.581 8.830 8.968 9.178 10.259 10.448 10.638 11.025 12.175 13.166 15.270 15.724 22.409 1.604 | 2.798 3.131 3.142 3.666 3.797 3.870 3.980 4.548 4.647 4.747 4.951 5.555 6.076 7.181 7.420 10.933 |
極性色譜柱 | ||
溶劑名稱 | tR/min | RART |
異辛烷 叔丁基甲基醚 甲基四氫呋喃 四氯化碳 丁酮 甲基異丙基酮 苯 乙酸丙酯 甲基異丁基酮 仲丁醇 | 1.691 1.807 1.856 1.957 1.966 2.053 2.063 2.217 2.267 2.303 2.488 2.988 3.094 3.126 3.511 3.561 3.653 3.821 3.833 4.017 4.207 4.295 4.303 4.875 5.005 5.041 5.069 5.275 5.275 6.437 7.108 7.735 7.892 8.068 8.533 8.848 | 0.033 0.076 0.094 0.131 0.135 0.167 0.171 0.228 0.246 0.260 0.328 0.513 0.552 0.564 0.707 0.725 0.759 0.822 0.826 0.894 0.964 0.997 1.000 1.212 1.260 1.273 1.284 1.360 1.360 1.790 2.039 2.271 2.329 2.394 2.566 2.683 |
續表
極性色譜柱 | ||
溶劑名稱 | tR/min | RART |
甲苯 正丙醇 二氧六環 乙酸丁酯 甲基丁基酮 異丁醇 異丙基苯 正戊醇 甲氧基苯 四氧化萘 | 9.156. 9.461 10.183 10.446 10.543 10.801 11.606 13.046 13.258 13.396 13.949 14.519 14.562 15.516 17.447 21.708 1.602 | 2.797 2.910 3.177 3.274 3.310 3.406 3.704 4.237 4.315 4.367 4.571 4.782 4.798 5.151 5.866 7.444 |
注:附表2、3中數據爲非極性的SPB-1柱(30m×0.32mm,1.0μm)和極性的HP-INNOWAX柱(30m×0.32mm,0.5μm)測定的結果。
25 附錄Ⅵ W 乙酰色氨酸測定法
本法系用紫外-可見分光光度法(吸收係數法)測定人血白蛋白供試品中的N-乙酰-DL-色氨酸含量。
25.1 測定法
用生理氯化鈉溶液將供試品蛋白質稀釋至5%,即爲供試品溶液.量取供試品溶液0.1ml,分別加入生理氯化鈉溶液0.3ml和0.3mol/L高氯酸溶液3.6ml,混勻;另取生理氯化鈉溶液0.4ml,加0.3mol/L高氯酸溶液3.6ml,混勻,作爲空白對照。室溫放置10分鐘,以每分鐘3500轉離。心20分鐘,取上清液在波長280nm處測定吸光度,用空白溶液調零點。按下式計算供試品中的N-乙酰-DL-色氨酸含量:

式中n爲供試品的稀釋係數;
P爲供試品的蛋白質含量,g/L。
26 附錄Ⅵ X 氰化物殘留量測定法
本法系依據在酸性條件下,溴化氰與吡啶聯苯胺發生顯色反應,採用紫外-可見分光光度法測定Hib多糖衍生物中溴化氰的含量。
26.1 試劑
(3)吡啶聯苯胺溶液 精密稱定聯苯胺0.5g,加入60%吡啶溶液50ml溶解,再加入2%鹽酸10ml,混勻。臨用前配製。
26.2 對照品溶液的製備
(1) 0.1mg/ml溴化氰對照品貯備液 稱定溴化氰10mg,加水定容至100ml(乙腈助溶)。臨用前配製。
(2)溴化氰對照品工作液(500ng/ml) 精密量取溴化氰對照品貯備液1ml,加水定容至200ml,混勻。
26.3 供試品溶液的製備
26.4 測定法
量取吡啶聯苯胺溶液2.0ml,加水2.0ml,混勻,20℃以下、暗處放置15分鐘後,在波長520nm處測定吸光度,作爲空白對照。
量取供試品溶液2.0ml、吡啶聯苯胺溶液2.0ml,混勻,20℃以下、暗處放置15分鐘後,在波長520nm處測定吸光度。
分別量取溴化氰對照品工作液0.1ml、0.2ml、0.4ml、0.6ml、0.8ml、1.0ml於試管中,每管依次加水1.9ml、1.8ml、1.6ml、1.4ml、1.2ml、1.0ml,加入吡啶聯苯胺溶液2.0ml,混勻,20℃以下、暗處放置15分鐘後,在波長520nm處測定吸光度。
26.5 結果計算
以對照品工作液的含量對其相應的吸光度作直線迴歸,求得直線迴歸方程,將供試品溶液的吸光度代入直線迴歸方程,求出供試品溶液中溴化氰的含量B(ng/ml)。
供試品中溴化氰的含量(ng/mg)=B/20
式中 B爲供試品溶液中溴化氰的含量,ng/ml;
27 附錄Ⅵ Y 碳二亞胺殘留量測定法
本法系依據二甲基巴比妥酸試液與碳二亞胺(EDAC)反應形成紫紅色絡合物,採用紫外-可見分光光度法測定碳二亞胺的含量。
27.1 試劑
(1)二甲基巴比妥酸試液 稱取二甲基巴比妥酸1g於16ml吡啶中,並加水至20ml.混勻,臨用現配。
(2)醋酸吡啶溶液 將等體積的冰醋酸和吡啶混勻製成,臨用現配。
(3)100μmol/L EDAC對照品貯備液 稱取0.0192gEDAC,以水溶解並定容至100ml,得1mmol/L溶液,量取1mmol/L溶液1ml於10ml量瓶,加水定容至刻度,即得100μmol/L EDAC對照品貯備液。臨用現配。(4)EDAC對照品工作液的製備 取100μmol/LEDAC對照品貯備液,用水分別稀釋至10μmol/L、20μmol/L、 40μmol/L、 60μmol/L、 80μmol/L, 即爲EDAC對照品工作液。
27.2 測定法
取供試品和EDAC對照品工作液各0.2ml,分別加入二甲基巴比妥酸試液1.8ml,另取水0.2ml作爲空白對照,同法操作,混勻各管,室溫暗處靜置30分鐘,分別加入醋酸吡啶溶液2.0ml,混勻後在波長599nm處測定吸光度(如試驗有干擾,在測吸光度前以每分鐘4000轉離心5分鐘)。
27.3 結果計算
以EDAC對照品溶液的濃度對其相應的吸光度作直線迴歸,求得直線迴歸方程。將供試品溶液的吸光度代入直線迴歸方程,求出含量,取其平均值。供試品EDAC殘留量(μmol/L)=供試品溶液EDAC的平均濃度×稀釋倍數
28 參考資料
- ^ [1] 國家藥典委員會.中華人民共和國藥典:2010年版:第一增補本[M].北京:中國醫藥科技出版社,2010.