2 附錄Ⅶ A 磷測定法
本法系將有機磷轉變爲無機磷後進行磷含量測定。磷酸根在酸性溶液中與鉬酸銨生成磷鉬酸銨,遇還原劑即生成藍色物質(三氧化鉬和五氧化鉬的混合物),稱之爲“鉬藍”,用比色法測定供試品中磷含量。
2.1 測定法
精密量取供試品適量(約含磷4~20μg)置試管中,加4滴硫酸(約0.08ml)加熱至炭化,再加2滴高氯酸(約0.06ml)消化至無色澄清,消化完全後稍置片刻,立即加水2ml,加0.04mol/L鉬酸銨溶液(稱取鉬酸銨5g,加水溶解並稀釋至100ml)0.4ml,混勻;加還原劑(稱取亞硫酸氫鈉6g、亞硫酸鈉1.2g、1-氨基-2-萘酚-4-磺酸0.1g,置棕色瓶中,加水至50ml,1周內使用)0.2ml,混勻;加水至6ml,15~20分鐘後,照紫外-可見分光光度法(2010年版藥典三部附錄Ⅱ A),在波長820nm處測定吸光度。
精密量取標準磷溶液(精密稱取乾燥至恆重的磷酸二氫鉀439.3mg,置100ml量瓶中,加水溶解並稀釋至刻度;再精密量取2ml,置100ml量瓶中,加水稀釋至刻度,即得每1ml含磷20μg的標準磷溶液)0.2ml、0.4ml、0.6ml、0.8ml、1.0ml,分別置試管中,各補加水至1ml,自“加4滴硫酸”起,同法操作,測定各管的吸光度。
以標準磷溶液的系列濃度對其相應的吸光度作直線迴歸,然後將供試品溶液的吸光度代入直線迴歸方程,求出其相應體積(ml)。
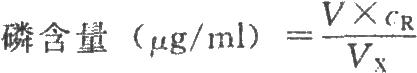
cR爲標準磷溶液的濃度,μg/ml;
VX爲供試品的體積,ml。
2.2 【附註】
(1)加高氯酸消化時,必要時可加1~2滴30%過氧化氫,但最後必須將過氧化氫除盡。
(3)測定A羣腦膜炎球菌多糖疫苗成品磷含量時,至少取3支安瓿溶解後混合備用。
3 附錄Ⅶ B 硫柳汞測定法
3.1 第一法 滴定法
本法系依據汞有機化合物經強酸消化成無機汞離子,與雙硫腙溶液形成橙黃色化合物,根據雙硫腙滴定液的消耗量,可計算出供試品中硫柳汞含量。
3.1.1 試劑
(1)雙硫腙滴定液 精密稱取雙硫腙50mg,置100ml量瓶中,加三氯甲烷溶解並稀釋至刻度,搖勻,作爲貯備液。
臨用前,精密量取貯備液2.5ml,置100ml量瓶中,加四氯化碳稀釋至刻度,搖勻,即得雙硫腙滴定液,保存於冷暗處。
(2)標準汞溶液 取置硫酸乾燥器中乾燥至恆重的氯化高汞約0.135g,精密稱定,置100ml量瓶中,加0.5mol/L硫酸溶解並稀釋至刻度,搖勻,即爲標準汞貯備液。
臨用前,精密量取標準汞貯備液適量,置100ml量瓶中,加0.5mol/L硫酸溶液稀釋至刻度,搖勻,即爲每1ml相當於50μg Hg的標準汞溶液。
3.1.2 測定法
(1)消化 精密量取供試品適量(約相當於含汞量50μg),置150ml圓底磨口燒瓶(附長40cm迴流管)中,加硫酸2ml、8.0mol/L硝酸溶液0.5ml混勻後,置電爐上加熱迴流15分鐘(或置於3cm×24cm試管中,加蓋置85~90℃水浴加熱1小時),冷卻後加水40ml,加20%鹽酸羥胺溶液5ml。
(2)滴定 用水40ml將上述消化後溶液分數次沖洗入125ml分液漏斗中,用雙硫腙滴定液滴定,開始時每次可加入2ml左右,以後逐漸減少至每次0.5ml,最後還可少至0.2ml。每次加入滴定液後,振搖10秒鐘,靜置分層,棄去四氯化碳層,繼續滴定,直至雙硫腙液的綠色不變,即爲終點。
(3)雙硫腙滴定液的標化 精密量取標準汞溶液1ml,置125ml分液漏斗中,加硫酸2ml,加水80ml和20%鹽酸羥胺溶液5ml,自“用雙硫腙滴定液滴定”起,同法操作。
按下式計算:
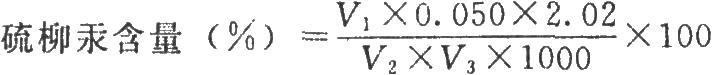
式中V1爲供試品消耗雙硫腙滴定液的體積,ml;
V3爲供試品的體積,ml;
0.050爲標準汞溶液的濃度,mg/ml,
【附註】
(1)抗毒素及免疫球蛋白供試品用水浴消化法滴定時會出現少量絮狀物,但不影響結果。
(2)可做限度測定。
3.2 第二法 原子吸收分光光度法
本法系依據有機汞在氧化條件下消化成無機汞離子,在氯化亞錫作用下將汞離子還原爲汞原子,採用原子吸收分光光度法測定供試品中汞含量,從而計算出硫柳汞含量。
3.2.1 試劑
(1)20%氯化亞錫溶液 稱取氯化亞錫20g,加鹽酸20ml,微熱溶解,冷卻室溫後加水稀釋至100ml。臨用時現配。
(2)稀硫酸 量取15ml硫酸(分析純),加水15ml,混勻。
(3)5%高錳酸鉀溶液 稱取5.0g高錳酸鉀(分析純),用水溶解並定容至100ml。煮沸10分鐘,靜置過夜,過濾。
(4)5%過硫酸鉀溶液 稱取5g過硫酸鉀,用水溶解並定容至100ml。臨用時現配。
(5)8%鹽酸羥胺溶液 稱取8g鹽酸羥胺,用水溶解並定容至100ml。
3.2.2 標準汞溶液的製備
3.2.3 供試品溶液的製備
取供試品適量,用水稀釋至所含汞濃度在標準曲線範圍內。
3.2.4 測定法
精密量取適量供試品和標準汞溶液,加稀硫酸4ml、硝酸1ml和5%高錳酸鉀溶液4ml,混勻,放置15分鐘後,加入5%過硫酸鉀溶液2ml,置約95℃加熱2小時,冷卻至室溫後,加8%鹽酸羥胺溶液2ml,加水至50ml後,取適量消化後供試品,並加入相應量的20%氯化亞錫溶液,照原子吸收分光光度法(2010年版藥典三部附錄Ⅱ B)室溫在波長253.7nm處測定吸光度,同時用水作空白對照。
3.2.5 結果計算
以標準汞溶液的濃度對其相應的吸光度作直線迴歸,相關係數不低於0.99,將供試品溶液的吸光度代入直線迴歸方程,即可得到供試品溶液汞含量。按下式計算供試品中的硫柳汞含量:
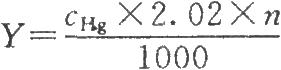
式中Y爲供試品中的硫柳汞含量,μg/ml;
cHg爲供試品溶液中的汞含量,ng/ml;
n爲供試品稀釋倍數。
4 附錄Ⅶ C 硫酸銨測定法
本法系依據硫酸銨被氫氧化鈉分解釋放出氨,並被硼酸吸收生成硼酸銨,用酸滴定液滴定。根據酸滴定液的消耗量可計算出供試品中硫酸銨含量。
4.1 供試品溶液的製備
除蛋白質方法同蛋白質含量測定(2010年版藥典三部附錄Ⅵ B 第一法)。
4.2 測定法
精密量取除蛋白質濾液10ml,置凱氏蒸餾器內,加4%氫氧化鈉溶液1ml,加少量水,照氮測定法(2010年版藥典三部附錄Ⅵ A)進行蒸餾、滴定,並將漓定的結果用空白試驗校正。
按下式計算:
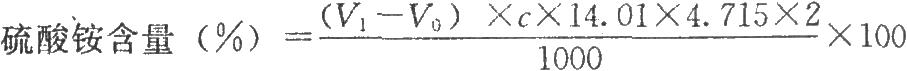
4.715爲常數(1g氮相當於4.715g硫酸銨);
14.01爲氮的相對原子質量。
5 附錄Ⅶ D 水分測定法
5.1 第一法(費休氏法)
5.1.1 A.容量滴定法
本法是根據碘和二氧化硫在吡啶和甲醇溶液中能與水起定量反應的原理以測定水分。所用儀器應乾燥,並能避免空氣中水分的侵入;測定操作宜在乾燥處進行。費休氏試液的製備與標定
(1)製備 稱取碘(置硫酸乾燥器內48小時以上)110g,置乾燥的具塞錐形瓶中,加無水吡啶160ml,注意冷卻,振搖至碘全部溶解後,加無水甲醇300ml,稱定重量,將錐形瓶置冰浴中冷卻,在避免空氣中水分侵入的條件下,通入乾燥的二氧化硫至重量增加72g,再加無水甲醇使成1000ml,密塞,搖勻,在暗處放置24小時。也可以使用穩定的市售卡爾-費休氏試液。市售的試液可以是不含吡啶的其他鹼化劑,不含甲醇的其他醇類等;也可以是單一的溶液或由兩種溶液混合而成。
(2)標定 精密稱取純化水10~30mg,用水分測定儀直接標定。
或精密稱取純化水10~30mg(視費休氏試液滴定度和滴定管體積而定),置乾燥的具塞玻璃瓶中,除另有規定外,加無水甲醇適量,在避免空氣中水分侵入的條件下,用本液滴定至溶液由淺黃色變爲紅棕色,或用電化學方法(如永停滴定法(二部附錄Ⅶ A)等]指示終點;另做空白試驗,按下式計算:
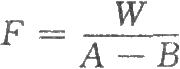
式中 F爲每1ml費休氏試液相當於水的重量,mg;
W爲稱取重蒸餾水的重量,mg;
測定法 精密稱取供試品適量,除另有規定外,溶劑爲無水甲醇,用水分測定儀直接測定。
或精密稱取供試品適量(約消耗費休氏試液1~5ml),置乾燥的具塞玻璃瓶中,加溶劑適量,在不斷振搖(或攪拌)下用費休氏試液滴定至溶液由淺黃色變爲紅棕色,或用電化學方法(如永停滴定法(2010年版藥典二部附錄Ⅶ A)等]指示終點;另做空白試驗,按下式計算:

式中A爲供試品所消耗費休氏試液的體積,ml;
F爲每1ml費休氏試液相當於水的重量,mg;
W爲供試品的重量,mg。
稱取供試品時,如供試品引溼性較強或毒性較大,可取適量置乾燥的容器中,密封(宜在通乾燥惰性氣體的手套操作箱中操作),精密稱定,用乾燥的注射器注入適量無水甲醇或其他適宜溶劑,精密稱定總重量,振搖使供試品溶解,測定該溶液的水分。洗淨並烘乾容器,精密稱定其重量,同時測定溶劑的水分。按下式計算:
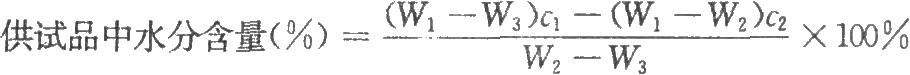
式中W1爲供試品、溶劑,和容器的重量,g;
W2爲供試品、容器的重量,g;
W3爲容器的重量,g;
此外,亦可將水分測定儀和市售卡氏乾燥爐聯用測定供試品水分。即將一定量的供試品在乾燥爐或樣品瓶中加熱,並用乾燥氣體將蒸發出的水分導入水分測定儀中測定。
5.1.2 B.庫侖滴定法
本法仍以卡爾-費休氏(Karl-Fischer)反應爲基礎,應用永停滴定法(2010年版藥典二部附錄Ⅶ A)測定水分。與容量滴定法相比,庫侖滴定法中滴定劑碘不是從滴定管加入,而是由含有碘離子的陽極電解液電解產生。一旦所有的水被滴定完全,陽極電解液中就會出現少量過量的碘,使鉑電極極化而停止碘的產生。根據法拉第定律,產生的碘的量與通過的電量成正比,因此可以用測量滴定過程中流過的總電量的方法測定水分總量。本法主要用於測定含微量水分(0.0001%~0.1%)的物質,特別適用於測定化學惰性物質如烴類、醇類和酯類中的水分。所用儀器應乾燥,並能避免空氣中水分的侵入;測定操作宜在乾燥處進行。
費休氏試液 按卡爾-費休氏庫侖滴定儀的要求配製或購置滴定液。本法無需標定滴定液。
測定法 先將系統中的水分預滴定除去,而後精密量取供試品適量(含水量約爲0.5~5mg),迅速轉移至陽極電解液中,用卡爾-費休氏庫侖滴定儀直接測定,以永停滴定法(2010年版藥典二部附錄Ⅶ A)指示終點,從儀器顯示屏上直接讀取供試品中水分的含量,其中每1mg水相當於10.72庫侖的電量。
5.2 第二法(甲苯法)
5.2.1 儀器裝置
如圖。A爲500ml的短頸圓底燒瓶;B爲水分測定管;C爲直形冷凝管,外管長40cm。使用前,全部儀器應清潔,並置烘箱中烘乾。
5.2.2 測定法
取供試品適量(約相當於含水量1~4ml),精密稱定,置A瓶中,加甲苯約200ml,必要時加入乾燥、潔淨的無釉小瓷片數片或玻璃珠數粒,將儀器各部分連接,自冷凝管頂端加入甲苯至充滿B管的狹細部分。將A瓶置電熱套中或用其他適宜方法緩緩加熱,待甲苯開始沸騰時,調節溫度,使每秒鐘餾出2滴。待水分完全餾出,即測定管刻度部分的水量不再增加時,將冷凝管內部先用甲苯沖洗,再用飽蘸甲苯的長刷或其他適宜方法,將管壁上附着的甲苯推下,繼續蒸餾5分鐘,放冷至室溫,拆卸裝置,如有水黏附在B管的管壁上,可用蘸甲苯的銅絲推下,放置使水分與甲苯完全分離(可加亞甲藍粉末少量,使水染成藍色,以便分離觀察)。檢讀水量,並計算成供試品的含水量(%)。
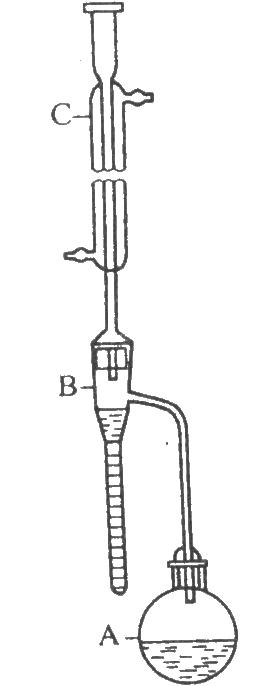
圖 甲苯法儀器裝置
6 附錄Ⅶ E 亞硫酸氫鈉測定法
本法系依據亞硫酸氫鈉與過量的碘反應,用硫代硫酸鈉滴定液滴定多餘的碘,根據硫代硫酸鈉滴定液的消耗量,可計算出供試品中亞硫酸氫鈉的含量。
6.1 測定法
精密量取供試品適量(約相當於含亞硫酸氫鈉量2.5mg),置具塞錐形瓶中,精密加入0.05mol/L碘溶液(稱取碘13.0g,加碘化鉀36g與水50ml溶解後,加鹽酸3滴與水適量使成1000ml,搖勻,用垂熔玻璃濾器濾過)20ml,放置5分鐘,沿瓶壁加入鹽酸溶液(5→10)2.0ml,搖勻。用硫代硫酸鈉滴定液(0.1mol/L)滴定至近終點時,加0.5%澱粉指示液約0.5ml,滴定至藍色消失,並將滴定的結果用空白試驗校正。
按下式計算:
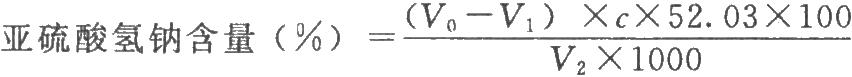
V2爲供試品的體積,ml;
6.2 【附註】
硫代硫酸鈉滴定液(0.1mol/L)的製備及滴定 稱取硫代硫酸鈉26g與無水碳酸鈉0.20g,加新沸過的冷水適量使溶解成1000ml,搖勻,放置1個月後濾過。
精密稱取在120℃乾燥至恆重的基準重鉻酸鉀0.15g,置碘瓶中,加水50ml使溶解,加碘化鉀2.0g,輕輕振搖使溶解,加稀硫酸(5.7→100) 40ml,搖勻,密塞,在暗處放置10分鐘後,加水250ml稀釋,用本液滴定至近終點時,加澱粉指示液(稱取可溶性澱粉0.5g,加水5ml攪勻後,緩緩傾人100ml沸水中,隨加隨攪拌,繼續煮沸2分鐘,冷卻,傾取上層清液。本液應臨用配製)3ml,繼續滴定至藍色消失而顯亮綠色,並將滴定的結果用空白試驗校正。每1ml硫代硫酸鈉滴定液相當於4.903mg的重鉻酸鉀。根據本液的消耗量與重鉻酸鉀的取用量,算出本液的濃度,即得。
7 附錄Ⅶ F 氫氧化鋁(或磷酸鋁)測定法
本法系依據過量的乙二胺四乙酸二鈉與鋁離子發生反應,再用鋅滴定液滴定剩餘的乙二胺四乙酸二鈉,根據鋅滴定液的消耗量,可計算出供試品中氫氧化鋁(或磷酸鋁)的含量。
7.1 測定法
精密量取供試品適量(約相當於含鋁1~10mg),置250ml錐形瓶中,加磷酸溶液(6→100)1.5ml,使完全溶解。必要時於水浴中加溫(難於溶解時尚可適當增加磷酸量)。精密加入乙二胺四乙酸二鈉滴定液(0.05mol/L) 10ml、醋酸-醋酸銨緩衝液(pH4.5)(稱取醋酸銨7.7g,加水50ml溶解後,加冰醋酸6ml與適量的水稀釋至100ml) 10ml,置沸水浴上加熱10分鐘,取出冷至室溫,加二甲酚橙指示液1ml,用鋅滴定液(0.025mol/L)進行滴定,當溶液由亮黃色變爲橙色,即爲終點,並將滴定的結果用空白試驗校正。
按下式計算:
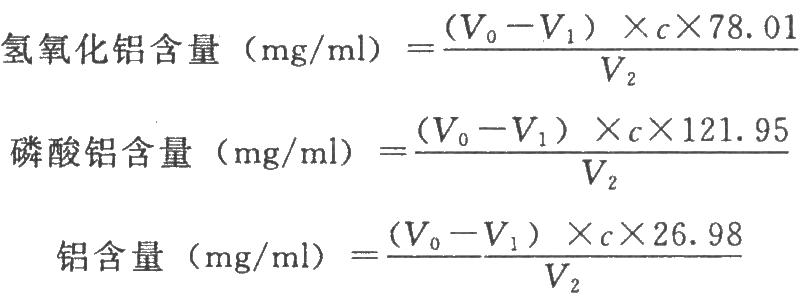
V1爲供試品消耗鋅滴定液的體積,ml;
c爲鋅滴定液的濃度,mol/L;
V2爲供試品的體積,ml;
78.01、121.95、26.98分別爲氫氧化鋁、磷酸鋁、鋁的分子量或相對原子質量。
7.2 【附註】
(1)鋅滴定液(0.05mol/L)的製備與滴定稱取硫酸鋅15g(相當於鋅約3.3g),加稀鹽酸(23.4→100) 10ml,適量水溶解並稀釋至1000ml,搖勻。精密量取本液25ml,加0.025%甲基紅的乙醇溶液1滴,滴加氨試液至溶液顯微黃色,加水25ml、氨-氯化銨緩衝液(pH10.0)10ml與鉻黑T指示劑少量,用乙二胺四乙酸二鈉滴定液(0.05mol/L)滴定至溶液由紫色變爲純藍色,並將滴定的結果用空白試驗校正。根據乙二胺四乙酸二鈉滴定液(0.05mol/L)的消耗量,算出本液的濃度,即得。
(2)鋅滴定液(0.025mol/L)的製備精密量取鋅滴定液(0.05mol/L) 100ml,加水準確稀釋至200ml,搖勻,即得。
(3)乙二胺四乙酸二鈉滴定液(0.05mol/L)的製備與滴定 稱取乙二胺四乙酸二鈉19g,加適量的水溶解並稀釋至1000ml,搖勻。取於約800℃灼燒至恆重的標準氧化鋅0.12g,精密稱定,加稀鹽酸(23.4→100) 3ml使溶解,加水25ml,加0.025%甲基紅的乙醇溶液1滴,滴加氨試液至溶液顯微黃色,加水25ml與氨-氯化銨緩衝液(pH10.0) 10ml,再加鉻黑T指示劑少量,用本液滴定至溶液由紫色變爲純藍色,並將滴定的結果用空白試驗校正。每1ml乙二胺四乙酸二鈉滴定液(0.05mol/L)相當於4.069mg的氧化鋅。根據本液的消耗量與氧化鋅的取用量,算出本液的濃度,即得。
8 附錄Ⅶ G 氯化鈉測定法
本法系用硝酸破壞供試品中的蛋白質後,再加入過量的硝酸銀,使供試品中的氯離子與硝酸銀完全反應,生成氯化銀沉澱析出,過量的硝酸銀用硫氰酸銨滴定液滴定,根據硫氰酸銨滴定液消耗的量,可計算出供試品中氯化鈉的含量。
8.1 測定法
精密量取供試品1.0ml,精密加入0.1mol/L硝酸銀溶液(稱取硝酸銀17.0g,加水溶解並稀釋至1000ml)5ml(若蛋白質含量較高者,加2ml飽和高錳酸鉀溶液),混勻,加8.0mol/L硝酸溶液10ml,加熱消化至溶液澄清,冷卻,加水50ml、8%硫酸鐵銨指示液1ml,用硫氰酸銨滴定液(0.05mol/L)滴定至溶液呈淡棕紅色,振搖後仍不褪色,即爲終點。將滴定的結果用空白試驗(可不消化)校正。
按下式計算:
氯化鈉含量(g/L)=(V0-Vx)×c×58.45
VX爲供試品消耗硫氰酸銨滴定液的體積,ml;
c爲硫氰酸銨滴定液濃度,mol/L;
8.2 【附註】
(1)硫氰酸銨滴定液(0.1mol/L)的製備及滴定 稱取硫氰酸銨8.0g,加水溶解並稀釋至1000ml,搖勻。精密量取硝酸銀滴定液(0.1mol/L)25ml,加水50ml、硝酸2ml與8%硫酸鐵銨指示液2ml,用本液滴定至溶液微顯淡棕紅色;經劇烈振搖後仍不褪色,即爲終點。根據本液的消耗量算出本液的濃度。
(2)硫氰酸銨滴定液(0.05mol/L)製備 精密量取硫氰酸銨滴定液(0.1mol/L) 100ml,加水準確稀釋至200ml,搖勻。
9 附錄Ⅶ H 枸櫞酸離子測定法
9.1 第一法 比色法
9.1.1 枸櫞酸鈉對照品溶液的製備
取經減壓乾燥至恆重的枸櫞酸鈉(C6H5Na3O7·2H2O)0.6g,精密稱定,置100ml量瓶中,加水溶解並稀釋至刻度,搖勻,精密量取5ml,置50ml量瓶中,用5%三氯乙酸稀釋至刻度,搖勻,即得。
9.1.2 供試品溶液的製備
精密量取供試品0.5ml與水4.5ml,加10%三氯乙酸溶液5ml,混勻,置60℃水浴加熱5分鐘,以每分鐘4000轉離心20分鐘,取上清液備用。
9.1.3 測定法
精密量取供試品溶液1ml,置25ml具塞試管中,精密加吡啶1.3ml,混勻,再精密加醋酸酐5.7ml,立即混勻並置31℃±1℃的水浴中,準確放置35分鐘後,照紫外-可見分光光度法(2010年版藥典三部附錄Ⅱ A),在波長425nm處測定吸光度。另精密量取枸櫞酸鈉對照品溶液0.25ml、0.50ml、0.75ml、1.0ml,分別置於具塞試管中,各精密加5%三氯乙酸溶液0.75ml、0.50ml、0.25ml、0.00ml(其相對應的枸櫞酸離子含量爲0.5mmol/L、1.0mmol/L、1.5mmol/L、2.0mmol/L),自“精密加吡啶1.3ml”起,同法操作。
以對照品溶液枸櫞酸離子濃度對其相應的吸光度作直線迴歸,求得直線迴歸方程,計算出供試品溶液中的枸櫞酸離子含量(mmol/L),再乘以供試品稀釋倍數(20),即爲供試品枸櫞酸離子含量(mmol/L)。
9.2 第二法 高效液相色譜法
照高效液相色譜法(2010年版藥典三部附錄Ⅲ B)測定。[1]
9.2.1 色譜條件
用苯乙烯-二乙烯基苯共聚物爲基質的陽離子交換色譜柱(H+),粒度9μm或8μm,內徑7.8mm,柱長300mm;柱溫爲50℃;流動相爲0.004mol/L硫酸溶液,流速爲每分鐘0.8ml,示差折光檢測器。
9.2.2 測定法
精密稱取經減壓乾燥至恆重的枸櫞酸鈉(C6H5Na3O7·2H2O)0.735g,置100ml量瓶中,用水溶解並稀釋至刻度,精密量取5.0ml、10.0ml、15.0ml,分別置25ml量瓶中,用水稀釋至刻度,搖勻,即得相對應的5.0mmol/L、10.0mmol/L、15.0mmol/L枸櫞酸離子對照品溶液。分別精密量取20μl,注入液相色譜儀,記錄色譜圖;另精密量取供試品溶液1ml,置15ml離心管中,精密加1.5%磺基水楊酸溶液1ml,混勻,室溫下以每分鐘2000轉離心10分鐘。取上清液,同法測定。
以對照品溶液的枸櫞酸離子濃度對其相應的峯面積作直線迴歸,求得直線迴歸方程,計算出供試品溶液枸櫞酸鈉含量(mmol/L),再乘以供試品稀釋倍數(2),計算出供試品枸櫞酸離子含量(mmol/L)。
9.2.3 【附註】
(1)根據供試品枸櫞酸離子含量,可適當調整枸櫞酸離子對照品溶液濃度。
(2)直線迴歸相關係數應不低於0.999。
(3)不同廠家的陽離子交換色譜柱(H+)的流速、流動相、柱溫等會有所不同,可根據色譜柱介紹對色譜條件進行適當調整。
9.3 第三法 高效液相色譜法
照高效液相色譜法(2010年版藥典三部附錄Ⅲ B)測定。
9.3.1 色譜條件與系統適用性試驗
色譜柱爲十八烷基硅烷鍵合硅膠填充色譜柱,柱長250mm,柱直徑4.6mm,粒度5μm。流動相爲18.2mmol/L磷酸鹽緩衝液,0.1%異丙醇溶液(pH2.0~2.5);柱溫:40℃;流速:每分鐘1.0ml;樣品池溫度:室溫;運行時間:50分鐘;紫外檢測器檢測波長:210nm。取5.0mmol/L枸櫞酸離子溶液20μl,注入色譜柱,記錄色譜圖,拖尾因子按枸櫞酸離子色譜峯測定應爲0.95~1.40。
9.3.2 測定法
精密稱取經減壓乾燥至恆重的枸櫞酸鈉(C6H5Na3O7·2H2O)0.735g,置100ml量瓶中,用超純水溶解並稀釋至刻度。精密量取5.0ml、10.0ml、15.0ml,分別置25ml量瓶中,用水稀釋至刻度,搖勻,即得相應的5.0mmol/L、10.0mmol/L、15.0mmol/L枸櫞酸離子對照品溶液。分別精密量取20μl,注入液相色譜儀,記錄色譜圖;另精密量取供試品溶液1ml,置15ml離心管中,精密加1.5%磺基水楊酸4ml,混勻。室溫靜置2小時以上,以每分鐘3000轉離心10分鐘,取上清液,同法測定。
以對照品溶液枸櫞酸離子濃度對其相應的峯面積作直線迴歸,求得直線迴歸方程。計算供試品溶液枸櫞酸離子含量(mmol/L),再乘以相應的供試品稀釋倍數(5),即爲供試品枸櫞酸離子含量(mmol/L)。
9.3.3 【附註】
(1)根據供試品枸櫞酸離子含量,可適當調整枸櫞酸離子對照品溶液濃度。
(2)根據供試品蛋白質濃度,可適當調整沉澱劑磺基水楊酸的加入量。
(3)直線迴歸相關係數應不低於0.999。
10 附錄Ⅶ I 鉀離子測定法
10.1 測定法
精密量取供試品2ml,置50ml量瓶中,用水稀釋至刻度,即爲供試品溶液。照火焰光度法(2010年版藥典三部附錄Ⅱ D)測定,在波長769nm處測定供試品溶液的發光強度。另精密稱取於110℃乾燥至恆重的氯化鉀56.0mg,置500ml量瓶中,用水溶解並稀釋至刻度,再精密量取該溶液1.0ml、2.0ml、3.0ml、4.0ml、5.0ml,分別置50ml量瓶中,用水稀釋至刻度,製成0.03mmol/L、0.06mmol/L、0.09mmol/L、0.12mmol/L、0.15mmol/L的系列標準鉀溶液,同法測定。
以系列標準鉀溶液的濃度對其相應的發光強度作直線迴歸,將供試品溶液發光強度代入直線迴歸方程,求得供試品溶液鉀離子濃度(mmol/L),再乘以供試品的稀釋倍數(25),計算出供試品鉀離子含量(mmol/L)。
11 附錄Ⅶ J 鈉離子測定法
11.1 測定法
精密量取供試品0.5ml,置50ml量瓶中,用水稀釋至刻度,即爲供試品溶液。照火焰光度法(2010年版藥典三部附錄Ⅱ D)測定,在波長589nm處測定供試品溶液的發光強度。另精密稱取於110℃乾燥至恆重的氯化鈉0.293g,置100ml量瓶中,用水稀釋至刻度,再精密量取該溶液0.9ml、1.1ml、1.3ml、1.5ml、1.7ml,分別置50ml量瓶中,用水稀釋至刻度,製成0.9mmol/L、1.1mmol/L、1.3mmol/L、1.5mmol/L、1.7mmol/L的系列標準鈉溶液,同法操作。
以系列標準鈉溶液的濃度對其相應的發光強度作直線迴歸,將供試品溶液發光強度代入直線迴歸方程,求得供試品溶液鈉離子濃度(mmol/L),再乘以供試品的稀釋倍數(100),計算出供試品鈉離子含量(mmol/L)。
12 附錄Ⅶ K 人血白蛋白鋁殘留量測定法
12.1 測定法
按表1精密量取供試品、100ng/ml標準鋁溶液(精密量取100μg/ml標準鋁溶液0.1ml,置100ml量瓶中,用0.15mol/L硝酸溶液稀釋至刻度),分別製備空白對照溶液、供試品溶液和標準鋁加供試品的混合溶液。照原子吸收分光光度法(2010年版藥典三部附錄Ⅱ B)測定,選擇鋁燈,測定波長爲309.3nm,狹縫爲0.7nm。按表2設置石墨爐的乾燥、灰化、原子化等爐溫程序,精密量取空白對照溶液、供試品溶液和標準鍋加供試品的混合溶液各30μl,分別注入儀器,讀數。
按下式計算:
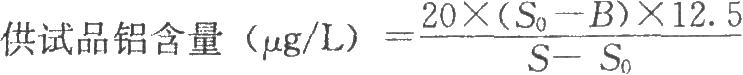
S0爲供試品溶液讀數;
S爲標準鋁加供試品的混合溶液讀數;
20爲標準鋁加供試品的混合溶液中標準鋁的含量,μg/L;
12.5爲供試品稀釋倍數。
12.2 【附註】
(1)供試品和標準鋁取量可根據儀器性能進行適當調整,使讀數在所用儀器可準確讀數範圍內。
(3)儘量避免使用玻璃容器。
13 附錄Ⅶ L 乾燥失重測定法
取供試品,混合均勻(如爲較大的結晶,應先迅速搗碎使成2mm以下的小粒),取約1g或各品種項下規定的重量,置與供試品相同條件下乾燥至恆重的扁形稱量瓶中,精密稱定,除另有規定外,在105℃乾燥至恆重。由減失的重量和取樣量計算供試品的乾燥失重。
供試品乾燥時,應平鋪在扁形稱量瓶中,厚度不可超過5mm,如爲疏鬆物質,厚度不可超過10mm。放入烘箱或乾燥器進行乾燥時,應將瓶蓋取下,置稱量瓶旁,或將瓶蓋半開進行乾燥;取出時,須將稱量瓶蓋好。置烘箱內乾燥的供試品,應在乾燥後取出置乾燥器中放冷,然後稱定重量。供試品如未達規定的乾燥溫度即融化時,應先將供試品於較低的溫度下乾燥至大部分水分除去後,再按規定條件乾燥。
當用減壓乾燥器(溫度通常爲室溫)或恆溫減壓乾燥器(除另有規定外,溫度爲60℃)時,除另有規定外,壓力應在2.67kPa (20mmHg)以下;乾燥器中常用的乾燥劑爲無水氯化鈣、硅膠或五氧化二磷;恆溫減壓乾燥器中常用的乾燥劑爲五氧化二磷,乾燥劑應保持在有效狀態。
14 附錄Ⅶ M 固體總量測定法
本法系在一定溫度下,使供試品的液體成分蒸發,用剩餘的固體成分計算供試品的固體總量。
14.1 測定法
14.1.1 第一法 105℃幹烤法
精密量取一定體積供試品於乾燥至恆重的適宜的玻璃稱量瓶中,置於烤箱中於105℃烘至恆重。
14.1.2 第二法 50℃幹烤法
精密量取一定體積供試品於乾燥至恆重的適宜的玻璃稱量瓶中,置於烤箱中於50℃烘至恆重。
按下式計算:

式中cX爲供試品的固體總量,g/ml;W爲供試品恆重後的重量,g;V爲供試品的體積,ml。
15 參考資料
- ^ [1] 國家藥典委員會.中華人民共和國藥典:2010年版:第一增補本[M].北京:中國醫藥科技出版社,2010.