2 附錄Ⅷ A 免疫印跡法
本法系以供試品與特異性抗體結合後,抗體再與酶標抗體特異性結合,通過酶學反應的顯色,對供試品的抗原特異性進行檢查。
2.1 試劑
(1)TG緩衝液 稱取三羥甲基氨基甲烷15.12g與甘氨酸72g,加水溶解並稀釋至500ml。4℃保存。
(2)EBM緩衝液 量取TG緩衝液20ml、甲醇40ml,加水稀釋至200ml。4℃保存。
(3)TTBS緩衝液 稱取三羥甲基氨基甲烷6.05g與氯化鈉4.5g,量取聚山梨酯80 0.55ml,加適量水溶解,用鹽酸調pH值至7.5,加水稀釋至500ml。4℃保存。
(4)底物緩衝液 稱取3,3'-二氨基聯苯胺鹽酸鹽15mg,加甲醇5ml與30%過氧化氫15μl,加TTBS緩衝液25ml使溶解,即得。臨用現配。
2.2 檢查法
照SDS-聚丙烯酰胺凝膠電泳法(2010年版藥典三部附錄Ⅳ C),供試品與陽性對照品上樣量應大於100ng。取出凝膠,切去凝膠邊緣,浸於EBM緩衝液中30分鐘。另取與凝膠同樣大小的厚濾紙6張、硝酸纖維素膜1張,用EBM緩衝液浸透。用半乾膠轉移儀進行轉移:在電極板上依次放上溼濾紙3張、硝酸纖維素膜1張、電泳凝膠、溼濾紙3張,蓋上電極板,按0.8mA/cm2硝酸纖維素膜恆電流轉移45分鐘。
取出硝酸纖維素膜浸入封閉液(10%新生牛血清的TTBS緩衝液,或其他適宜封閉液)封閉60分鐘。棄去液體,加入TTBS緩衝液10ml,搖動加入適量的供試品抗體(參考抗體使用說明書的稀釋度稀釋),室溫過夜。硝酸纖維素膜用TTBS緩衝液淋洗1次,再用TTBS緩衝液浸洗3次,每次8分鐘。棄去液體,再加入TTBS緩衝液10ml,搖動加入適量的生物素標記的第二抗體,室溫放置40分鐘。硝酸纖維素膜用TTBS緩衝液淋洗1次,再用TTBS緩衝液浸洗3次,每次8汾鍾。棄去液體,更換TTBS緩衝液10ml,搖動,加入適量的親和素溶液和生物素標記的辣根過氧化物酶溶液,室溫放置60分鐘。硝酸纖維素膜用TTBS緩衝液淋洗1次,再用TTBS緩衝液浸洗4次,每次8分鐘。棄去液體,加入適量底物緩衝液,置於室溫避光條件下顯色,顯色程度適當時水洗終止反應。
2.3 結果判定
3 附錄Ⅷ B 免疫斑點法
本法系以供試品與特異性抗體結合後,抗體再與酶標抗體特異性結合,通過酶學反應的顯色,對供試品的抗原特異性進行檢查。
3.1 試劑
(1) TG緩衝液 精密稱取三羥甲基氨基甲烷15.12g與甘氨酸72g,加水溶解並稀釋至500ml。4℃保存。
(2) EBM緩衝掖 量取TG緩衝液20ml、甲醇40ml,加水稀釋至200ml。4℃保存。
(3) TTBS緩衝液 稱取三羥甲基氨基甲烷6.05g、氯化鈉4.5g,吸取聚山梨酯80 0.55ml,加適量水溶解,用鹽酸調pH值至7.5,加水稀釋至500ml。4℃保存。
(4)底物緩衝液 稱取3,3'-二氨基聯苯胺鹽酸鹽(DAB)15mg,取甲醇5ml、30%過氧化氫15μl,溶於25ml TTBS緩衝液中。用前配製。
3.2 檢查法
取硝酸纖維素膜,用EBM緩衝液浸泡15分鐘,將供試品、陰性對照品(可用等量的人白蛋白)及陽性對照品點在膜上,上樣量應大於10ng。室溫乾燥60分鐘。取出硝酸纖維素膜,浸入封閉液(10%新生牛血清的TTBS緩衝液,或其他適宜的封閉液)封閉60分鐘。棄去液體,加入TTBS緩衝液10ml,搖動加入適量的供試品抗體(參考抗體使用說明書的稀釋度稀釋),室溫過夜。硝酸纖維素膜用TTBS緩衝液淋洗1次,再用TTBS緩衝液浸洗3次,每次8分鐘。棄去液體,更換TTBS緩衝液10ml,搖動加入適量的生物素標記的第二抗體,室溫放置40分鐘。硝酸纖維素膜用TTBS緩衝液淋洗1次,再用TTBS緩衝液浸洗3次,每次8分鐘。棄去液體,更換TTBS緩衝液10ml,搖動加入適量的親和素溶液和生物素標記的辣根過氧化物酶溶液,室溫放置60分鐘。硝酸纖維素膜用TTBS緩衝液淋洗1次,再用TTBS緩衝液浸洗4次,每次8分鐘。棄去液體,加入適量底物緩衝液置於室溫避光條件下顯色,顯色程度適當時水洗終止反應。
3.3 結果判定
4 附錄Ⅷ C 免疫雙擴散法
本法系在瓊脂糖凝膠板上按一定距離打數個小孔,在相鄰的兩孔內分別加入抗原與抗體,若抗原、抗體互相對應,濃度、比例適當,則一定時間後,在抗原與抗體孔之間形成免疫複合物的沉澱線,以此對供試品的特異性進行檢查。
4.1 供試品溶液的製備
4.2 試劑
(1)0.5%氨基黑染色劑 稱取氨基黑10B0.5g,加甲醇50ml、冰醋酸10ml與水40ml的混合液,溶解,即得。
(2)脫色液 量取乙醇45ml、冰醋酸5ml與水50ml混合均勻,即得。
4.3 檢查法
將完全溶脹的1.5%瓊脂糖溶液傾倒於水平玻板上(每平方釐米加0.19ml瓊脂糖),凝固後,按下圖打孔,直徑3mm,孔距3mm(方陣型)。根據需要確定方陣型圖數量。中央孔加入抗血清,周邊孔加入供試品溶液,並留1孔加入相應陽性對照血清。每孔加樣20μl,然後置水平溼盒中,37℃水平擴散24小時。用生理氯化鈉溶液充分浸泡瓊脂糖凝膠板,以除去未結合蛋白質。將浸泡好的瓊脂糖凝膠板放入0.5%氨基黑溶液中染色。用脫色液脫色至背景無色,沉澱線呈清晰藍色爲止。用適當方法保存或複製圖譜。

圖 方陣型
4.4 結果判定
5 附錄Ⅷ D 免疫電泳法
本法系將供試品通過電泳分離成區帶的各抗原,然後與相應的抗體進行雙相免疫擴散,當兩者比例合適時形成可見的沉澱弧。將沉澱弧與已知標準抗原、抗體生成的沉澱弧的位置和形狀進行比較,即可分析供試品中的成分及其性質。
5.1 試劑
(1)巴比妥緩衝液(pH8.6) 稱取巴比妥4.14g與巴比妥鈉23.18g,加適量水,加熱使溶解,冷卻至室溫,再加疊氮鈉0.15g,加水使溶解成1500ml。
(2)0.5%氨基黑染液 稱取氨基黑10B 0.5g.加甲醇50ml、冰醋酸10ml與水40ml的混合液,溶解。
(3)1.5%瓊脂糖溶液 稱取瓊脂糖1.5g,加水50ml與巴比妥緩衝液50ml,加熱使溶脹完全。
(4)脫色液 量取乙醇45ml、冰醋酸5ml與水50ml,混合均勻。
(5)溴酚藍指示液 稱取溴酚藍50mg,加水使溶解成100ml。
5.2 對照品
正常人血清或其他適宜的對照品。
5.3 供試品溶液的製備
5.4 檢查法
將1.5%瓊脂糖溶液傾倒於大小適宜的水平玻板上,厚度約3mm,靜置,待凝膠凝固成無氣泡的均勻薄層後,於瓊脂糖凝膠板負極1/3處的上下各打1孔,孔徑3mm,孔距10~15mm。測定孔加供試品溶液10μl和溴酚藍指示液1滴,對照孔加正常人血清或人血漿10μl和溴酚藍指示液1滴。用3層濾紙搭橋和巴比妥緩衝液(電泳緩衝液)接觸,100V恆壓電泳約2小時(指示劑遷移到前沿)。電泳結束後,在兩孔之間距離兩端約3~5mm處挖寬3mm槽,向槽中加入血清抗體或人血漿抗體,槽滿但不溢出。放溼盒中37℃擴散24小時。擴散完畢後,用生理氯化鈉溶液充分浸泡瓊脂糖凝膠板,以除去未結合蛋白質。將浸泡好的瓊脂糖凝膠板放入0.5%氨基黑溶液染色,再用脫色液脫色至背景基本無色。用適當方法保存或複製圖譜。與對照品比較,供試品的主要沉澱線應爲待測蛋白質。
5.5 注意事項
6 附錄Ⅷ E 肽圖檢查法
本法系通過蛋白酶或化學物質裂解蛋白質後,採用適宜的分析方法鑑定蛋白質一級結構的完整性和準確性。
6.1 第一法 胰蛋白酶裂解-反相高效液相色譜法
照高效液相色譜法(2010年版藥典三部附錄Ⅲ B)測定。
6.1.1 色譜條件
以蛋白質與多肽分析用辛烷基硅烷鍵合硅膠或十八烷基硅烷鍵合硅膠爲填充劑;柱溫爲30℃±5℃,對照品與供試品保存溫度爲2~8℃;以0.1%三氟乙酸的水溶液爲流動相A液,以0.1%三氟乙酸的乙腈溶液爲流動相B液,流速爲每分鐘1ml,梯度洗脫70分鐘(A液從100%~30%,B液從0~70%),檢測波長爲214nm。
6.1.2 檢查法
取供試品溶液及對照品溶液(均爲每1ml中含1mg的溶液,如供試品和對照品濃度不夠,則應濃縮至相應的濃度),分別用1%碳酸氫銨溶液充分透析,按1:50 (mg/mg)加入胰蛋白酶溶液[取甲苯磺酰苯丙氨酰氯甲酮處理過的(或序列分析純)胰蛋白酶適量,加1%碳酸氫銨溶液溶解,製成每1ml中含0.1mg的溶液]到供試品溶液與對照品溶液中,於37℃保溫16~24小時後,按1: 10加入50%醋酸溶液,以每分鐘10000轉離心5分鐘(或用0.45μm濾膜濾過),精密量取上清液100μl,分別注入液相色譜儀,梯度洗脫,記錄色譜圖。將供試品溶液的圖譜與對照品溶液的圖譜進行比較,即得。
6.2 第二法 溴化氰裂解法
檢查法 取供試品與對照品適量(約相當於蛋白質50μg),用水透析16小時,冷凍乾燥,加溴化氰裂解液(稱取溴化氰0.3g,加甲酸(70→100)1ml使溶解]20μl溶解,室溫放置24小時,裂解物加水180μl,再冷凍乾燥。凍乾的裂解物用水復溶至適當濃度.照SDS-聚丙烯酰胺凝膠電泳法(附錄Ⅳ C)(膠濃度20%)進行電泳,用銀染法染色。
將供試品圖譜與對照品圖譜進行比較,即得。
7 附錄Ⅷ G A羣腦膜炎球菌多糖分子大小測定法
7.1 第一法 測磷法(仲裁法)
本法用於測定細菌莢膜多糖在色譜柱中的分配係數(KD)和多糖在規定KD值以前的回收率。
7.1.1 試劑
(1)流動相 稱取氯化鈉11.7g、疊氮鈉0.1g,加水使溶解成1000ml,混勻,用0.1mol/L氫氧化鈉溶液調pH值至7.0。
(2)藍色葡聚糖2000溶液 稱取藍色葡聚糖200020mg,加流動相使溶解成10ml。
(3)維生素B12溶液 稱取10mg維生素B12,加流動相使溶解成10ml。
7.1.2 色譜柱的製備
取瓊脂糖4B凝膠或瓊脂糖CL-4B凝膠約200ml,加流動相400ml充分攪拌,放置約1小時使其沉澱,傾去上層含懸浮顆粒的懸液。如此反覆3~5次後,加流動相200ml,混勻,抽去凝膠中的空氣,裝於1.5cm×90cm色譜柱中,約87cm高,用流動相洗脫,流速爲每小時15~20ml,以2~3倍柱牀體積的流動相洗脫(約500ml),使柱牀平衡。
7.1.3 色譜柱的標定
取藍色葡聚糖2000溶液1ml,加至已平衡的色譜柱中,以流動相洗脫,流速每小時15~20ml,用組分收集器收集洗脫液,每管收集3~5ml,照紫外-可見分光光度法(附錄Ⅱ A),在波長260nm處測定各管洗脫液的吸光度,以吸光度爲縱座標,洗脫液體積(ml)爲橫座標分別作圖,波長260nm處的峯頂洗脫液體積爲空流體積VO。
量取維生素B12溶液1ml,自“加至已平衡的色譜柱中”起,同法操作,370nm波長處的峯頂洗脫液體積爲柱牀體積Vi。
7.1.4 測定法
取供試品約1ml(含多糖抗原3~5mg.如爲凍乾製品可用流動相溶解),加至已標定的色譜柱中,用流動相洗脫,用組分收集器收集洗脫液,每管收集5ml,照磷測定法(2010年版藥典三部附錄Ⅶ A)測定每管洗脫液的磷含量。以供試品每管洗脫液的磷含量爲縱座標,洗脫液體積(ml)爲橫座標作圖,主峯峯頂洗脫液體積爲Ve。
按下式計算:

式中 KD爲供試品分配係數;
Ve爲供試品洗脫液體積,ml;
VO爲空流體積,ml;
Vi爲柱牀體積,ml。
計算供試品在KD值<0.5的多糖回收率:

式中RX爲KD值<0.5供試品的多糖回收率,%;
AX爲供試品在KD值<0.5各管洗脫液的磷含量之和;
A爲供試品所有管洗脫液的磷含量之和。
7.2 第二法 儀器法
試劑與色譜柱的製備同第一法。
7.2.1 色譜柱的標定
量取藍色葡聚糖2000溶液1ml與維生素B12溶液0.2ml;混勻後加至已平衡的色譜柱中,以流動相洗脫,流速每小時15~20ml,檢測波長206nm,用組分收集器收集洗脫液,記錄色譜圖,色譜圖中,第一峯爲籃色葡聚糖2000峯,峯頂的洗脫液體積爲空流體積VO;第二峯爲維生素B12峯,峯頂的洗脫液體積爲柱牀體積Vi。
7.2.2 測定法
取供試品約1ml(含多糖抗原3~5mg,如爲凍乾製品可用流動相溶解),加至已標定的色譜柱中,用流動相洗脫,流速爲每小時15~20ml,檢測波長206nm,用組分收集器收集洗脫液,記錄色譜圖,即得。按下式計算:

式中 KD爲供試品分配係數;
Ve爲供試品洗脫液體積,ml;
VO爲空流體積,ml;
Vi爲柱牀體積,ml。
計算供試品在KD值<0.5的多糖回收率:

式中RX爲KD值<0.5供試品的多糖回收率,%;
AX爲供試品在KD值<0.5的色譜圖面積;
At爲供試品色譜圖總面積。
7.3 【附註】
過柱操作在10~20℃進行。
8 附錄Ⅷ H 傷寒Vi多糖分子大小測定法
本法用於測定細菌莢膜多糖在色譜柱中的分配係數(KD)和多糖在規定KD值以前的回收率。
8.1 試劑、色譜柱的製備與色譜柱標定
同附錄Ⅷ G 第二法。
8.2 測定法
取供試品約1ml(含多糖抗原3~5mg),加至已標定的色譜柱中,用流動相洗脫,流速爲每小時15~20ml,用組分收集器收集洗脫液,每管3~5ml。照O-乙酰基測定法(2010年版藥典三部附錄Ⅵ F),測定每管洗脫液中O-乙酰基的含量,求出O-乙酰基含量最高時的洗脫體積,即爲多糖主峯峯頂洗脫體積Ve。
按下式計算:

式中KD爲供試品分配係數;
Ve爲供試品洗脫液體積,ml;
VO爲空流體積,ml;
Vi爲柱牀體積,ml。
計算供試品在KD值≤0.25的多糖回收率:
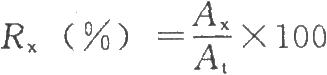
式中RX爲KD值≤0.25供試品的多糖回收率,%;
AX爲供試品在KD值≤0.25各管洗脫液等體積合併液的O-乙酰基含量;
8.3 【附註】
過柱操作在10~20℃進行。
9 附錄Ⅷ I 牛血清白蛋白殘留量測定法
9.1 供試品溶液的製備
供試品如爲凍幹劑型,檢測前應按標示量復溶後混勻,室溫靜置30分鐘,檢測前應再次混勻。供試品如爲液體劑型可直接用於檢測。
9.2 干擾試驗
製備溶液Ⅰ(供試品倍比稀釋)、溶液Ⅱ(供試品和30ng/ml的內控標準品等量混合)和溶液Ⅲ(30ng/ml的內控標準品倍比稀釋)。當供試品溶液BSA含量高於試劑盒測定範圍中點時,則2倍稀釋後製備溶液Ⅰ和溶液Ⅱ。溶液Ⅰ、溶液Ⅱ可倍比稀釋測定,溶液Ⅲ應多孔測定(至少10孔以上),並在試驗間均勻添加。按測定法操作,分別測定溶液Ⅰ、溶液Ⅱ、溶液Ⅲ的BSA含量,溶液Ⅰ與溶液Ⅱ的含量之差應在溶液Ⅲ含量測定值的95%可信區間內,表明供試品不會對該檢測法產生干擾作用。
9.3 測定法
按試劑盒介紹進行,並採用試劑盒提供的供試品稀釋液稀釋供試品,供試品應至少進行2個稀釋度測定,每個稀釋度做雙孔平行測定。試劑盒標準品的吸光度、內控參考品測定值、標準品線性相關係數、雙孔測定吸光度均應在試劑盒要求範圍內,試驗有效。以標準品溶液的濃度對其相應的吸光度作直線迴歸,將供試品的吸光度代入直線迴歸方程,再乘以稀釋倍數,計算出供試品中BSA含量。
9.4 【附註】
(1)當同一供試品的低稀釋度吸光度明顯低於高稀釋度吸光度時,可能存在HOOK效應或操作失誤,需重試或調整稀釋倍數進行檢測。
(2)測定BSA含量的容器具應專用,防止實驗室中BSA污染。
10 附錄Ⅷ J b型流感嗜血桿菌結合疫苗多糖含量測定法
本法系依據可溶性糖經無機酸處理脫水產生糖醛(戊糖)或糖醛衍生物,生成物能與酚類化合物縮合生成有色物質,以此測定多糖的含量。
10.1 試劑
(1)0.1%三氯化鐵鹽酸溶液 準確稱取三氯化鐵(FeCl3·6H2O)0.1g,放入清潔的試劑瓶內,加鹽酸100ml,待溶解後置2~8℃冰箱保存。
(2)地衣酚(3,5-二羥基甲苯)[1]乙醇溶液 稱取地衣酚1g,放入10ml量瓶中,加95%乙醇至10ml。臨用前配製。
(3) 25μg/ml核糖對照品溶液 稱取D-核糖1.25mg,置50ml量瓶中,加水溶解並稀釋至刻度。
10.2 測定法
量取1ml水,加入5ml 0.1%三氯化鐵鹽酸溶液,混勻後再加入0.4ml地衣酚乙醇溶液,混勻。水浴5分鐘後置冰浴,在波長670nm處測定吸光度,作爲空白對照。
先將供試品用水稀釋至核糖含量不高於25μg/ml,作爲供試品溶液,量取1.0ml自“加入5ml 0.1%三氯化鐵鹽酸溶液”起,同法操作。
分別取核糖對照品溶液0.1ml、0.2ml、0.4ml、0.6ml、0.8ml、1.0ml於10ml試管中,每管依次加水0.9ml、0.8ml、0.6ml、0.4ml、0.2ml、0ml,自“加入5ml 0.1%三氯化鐵鹽酸溶液”起,同法操作。
10.3 結果計算
以核糖對照品溶液的濃度對其相應的吸光度作直線迴歸,求得直線迴歸方程。將供試品溶液的吸光度代入直線迴歸方程,求出供試品溶液的核糖含量。
供試品多糖含量(μg/ml)=a×n/0.41
式中a爲供試品溶液的核糖含量,μg/ml;
n爲供試品稀釋倍數。
11 附錄Ⅷ K 己二酰肼含量測定法
本法系依據在四硼酸鈉存在的條件下,己二酰肼(ADH)中的氨基基團能與三硝基苯磺酸(TNBS)發生顯色反應,採用紫外-可見分光光度法測定b型流感嗜血桿菌多糖衍生物中己二酰肼的含量。
11.1 試劑
(1)己二酰肼對照品貯備液(1mg/ml) 精密稱定己二酰肼0.100g,加水定容至100ml,於-20℃保存。
(2)己二酰肼對照品工作液(20μg/ml) 精密量取ADH對照品貯備液0.2ml,加水定容至10ml。
(3)5%四硼酸鈉溶液 稱取四硼酸鈉(Na2B4O7·10H2O) 47.35g[1],加水定容至500ml,於室溫保存。
(4) 3% TNBS溶液 量取TNBS 5ml,加水定容至50ml,於-20℃保存。
11.2 測定法
量取5%四硼酸鈉溶液1.0ml,加水1ml,混勻,再加入3% TNBS溶液0.3ml,混勻,於室溫放置15分鐘,在波長500nm處測定吸光度,作爲空白對照。先將供試品用水稀釋至己二酰肼濃度不高於20μg/ml,作爲供試品溶液,然後取1.0ml,加入5%四硼酸鈉溶液1.0ml,自“加入3% TNBS溶液0.3ml”起同法操作。
分別取己二酰肼對照品工作液0.2ml、0.4ml、0.6ml、0.8ml、1.0ml於試管中,每管依次加水0.8ml、0.6ml、0.4ml、0.2ml、0ml,加入5%四硼酸鈉溶液1.0ml,自“加入3% TNBS溶液0.3ml”起同法操作。結果計算 以己二酰肼對照品工作液的濃度對其相應的吸光度作直線迴歸,求得直線迴歸方程,將供試品溶液的吸光度代入直線迴歸方程,求出供試品溶液的己二酰肼含量,根據稀釋倍數計算供試品的己二酰肼含量。
12 附錄Ⅷ L 高分子結合物含量測定法
本法系利用高分子結合物、低分子結合物及遊離多糖在不同乙醇濃度下,沉澱分離,採用紫外-可見分光光度法測定磷含量,計算高分子結合物的含量。
12.1 試劑
(1) 5mol/L氯化鈉溶液 精密稱定氯化鈉29.22g,加水溶解並稀釋至100ml,室溫保存。
(2) 1.5mol/L硫酸 於1體積98%的硫酸中加入11體積的水,混勻。
(3) 2.5%鉬酸銨 稱取鉬酸銨2.65g[1],加水溶解並稀釋至100ml。
(4)10%抗壞血酸 稱取抗壞血酸10g,加水溶解並稀釋至100ml。
(6)產色試劑 水、1.5mol/L硫酸、2.5%鉬酸銨、10%抗壞血酸,按2:1:1:1體積比混合配製。
(7)80μg/ml磷對照品貯備液 精密稱定經100℃乾燥的磷酸氫二鈉0.3665g或磷酸二氫鉀0.3509g[1],加水500ml、5mol/L硫酸溶液10ml溶解,補加水至1000ml。臨用時,將貯備液做20倍稀釋,即爲4μg/ml磷對照品工作液。
(8)1.0mol/L氫氧化鈉溶液 稱取4g氫氧化鈉,加水溶解並稀釋至100ml。
12.2 供試品溶液的製備
(1)分步沉澱 原液用生理氯化鈉溶液稀釋至多糖含量20~28μg/ml或成品疫苗3ml,加入5mol/L氯化鈉溶液0.75ml,混勻後加入無水乙醇15ml,於-20℃冰箱放置72~96小時,以每分鐘8000轉4℃離心90分鐘,吸取上清液爲供試品溶液2;於沉澱中加入50%乙醇溶液0.5ml,加玻璃珠,混合後室溫放置1小時;再加入50%乙醇溶液1.5ml,混合後室溫放置2小時,然後以每分鐘8000轉8℃離心l小時,吸取1.8ml上清液爲供試品溶液3;沉澱再加入1.0mol/L氫氧化鈉溶液0.5ml,混合後室溫放置1小時,加水1.25ml,作爲供試品溶液4。
(2)取多糖含量20~28μg/ml的原液或成品疫苗1.0ml爲供試品1。
(3)供試品溶液的礦化 分別量取1.0ml供試品1、1.5ml供試品溶液2、0.7ml供試品溶液3、0.5ml供試品溶液4各2份;分別加入礦化試劑0.15ml,置150℃乾燥1小時,然後升溫至180℃乾燥30分鐘,再升溫至250℃乾燥1小時。
12.3 測定法
量取水1.95ml,加礦化試劑50μl後加2.0ml產色試劑,混勻後置37℃水浴2小時,在波長825nm處測定吸光度,作爲空白對照。
於礦化好的供試品溶液中加水1.85ml,加產色試劑2.0ml,混勻後置37℃水浴2小時,在波長825nm處測定吸光度。
分別量取磷對照品工作液0.1ml、0.2ml、0.4ml、0.8ml、1.0ml於試管中,每管依次加水1.85ml、1.75ml、1.55ml、1.15ml、0.95ml;然後每管分別加入礦化試劑50μl後加2.0ml產色試劑,混勻後置37℃水浴2小時,在波長825nm處測吸光度。
12.4 結果計算
以磷對照品溶液的濃度對其相應的吸光度作直線迴歸,求得直線迴歸方程。將供試品溶液的吸光度代入直線迴歸方程,求出磷含量。
供試品磷含量(μg/ml)分別爲:
P1= (A1×3)/1.0
P2-(A2×18. 75)/1.5
P3= (A3×2.0)/0.7
P4= (A4×2.0)/0.5-(P3×10)/100
試驗有效性 80%≤P1/(P2+P3+P4)≤120%
供試品高分子結合物含量(%)=P4/(P2+P3+P4)×100
供試品遊離多糖含量(%)=[1-P4/(P2+P3+P4)]×100
式中 P1、P2、P3、P4爲供試品1,供試品溶液2,供試品溶液3,供試品溶液4的磷含量;A1、A2、A3、A4爲供試品1,供試品2,供試品3,供試品4中取樣礦化後的磷含量。
13 參考資料
- ^ [1] 國家藥典委員會.中華人民共和國藥典:2010年版:第一增補本[M].北京:中國醫藥科技出版社,2010.