3 概述
腸病性肢端皮炎(acrodermatitis enteropathica)是一種少見的常染色體隱性遺傳性疾病,以腔口周圍、四肢末端之皮炎、腹瀉、禿髮爲其臨牀特徵。本病常於1歲左右開始發病,鋅治療有效,嚴重病例不治可致死亡。本病於1943年首先由Danbolt和Closs描述,此後國內外有許多報道。
6 別名
acrodermatitis enteropathica syndrome;Brandt綜合徵;Danbolt-Closs綜合徵;布蘭特氏綜合徵;布蘭特綜合徵;腸病性肢皮炎;腸源性肢端皮炎綜合徵;當-克二氏綜合徵;非典型性連續性肢端皮炎;慢性腸源性肢端皮炎
10 病因
腸病性肢端皮炎的病因學說曾有多種推測,如腸道吸收功能紊亂,基本氨基酸吸收不良導致蛋白質和脂肪代謝障礙,母乳中缺乏一種未知因子,色氨酸代謝異常產生對皮膚和腸道上皮有毒性作用的代謝產物,系統性白念珠菌感染等,均因缺乏足夠證據而被否定。部分患者有明顯家族史,雙親爲近親者發病率高,認爲本病屬常染色體隱性遺傳。現認爲本病爲一遺傳性鋅缺乏病,存在胃腸道鋅吸收障礙。究其原因是腸道中一種胰腺分泌的鋅結合因子(可能是前列腺素E2)水平不足所致。
12 腸病性肢端皮炎的臨牀表現
起病於嬰幼兒期,主要見於出生後1歲內的幼嬰,斷奶前後發病率最高,爲慢性多形性皮炎表現,皮損初起爲小皰,皰液初透明,以後因繼發細菌或真菌,尤其是白色念珠菌感染而混濁,皮損互相融合結痂,形成不同大小斑片,周圍繞以紅暈,有的皮損呈銀屑病樣或苔蘚樣外觀。水皰尼氏徵陰性。病變部位主要在腔口周圍(口腔、眼周、鼻孔、肛周、會陰、生殖器等)、肢端(手指、足趾、足跟)及四肢摩擦部位(肘關節、膝蓋)(圖1)。在所有病例均可見脫髮現象發生,一般於皮疹出現前開始,先是受機械刺激的頭後,然後是頭的兩側。頭髮瀰漫性稀少,細軟無光澤。眉毛、睫毛也可脫落。胃腸道症狀以腹瀉爲主,排出酸臭的水樣便,但約10%的病人無腸道症狀。多數患者出現指甲變形、萎縮和脫落、甲溝炎等。嚴重者出現口角糜爛、口腔炎、結合膜炎、鼻炎、畏光、角膜混濁。皮疹惡化時有精神不佳、易怒、面部傾斜、低頭、智力障礙。
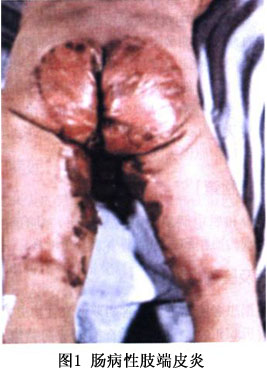
病程呈波動性,有的在青春發育期症狀改善,妊娠期加劇,當分娩或流產後則症狀又可消失,可能是由於食物中含鋅量的變化,以及當機體在生長發育期、妊娠期或感染性疾病時所需鋅量的改變所致。在較嚴重的病例,身體生長和性成熟障礙,並伴精神壓抑,可出現喪失人格,嚴重病例不治可致死亡。
13 實驗室檢查
血清鋅水平降低(正常值9.18~19.89μmol/L);毛髮、指甲、皮膚和尿鋅濃度因正常變動範圍大,故不可靠。鹼性磷酸酶是含鋅金屬酶,隨血鋅缺乏而降低,當肝功能正常,低鹼性磷酸酶活性伴低血鋅可作爲鋅缺乏佐證。其它有皮膚粘膜損害處、尿和糞中檢出白念珠菌、貧血、低蛋白和低白蛋白血癥、脂肪痢等。
14 輔助檢查
組織病理:皮膚活檢無特異性,呈現亞急性皮炎組織病理改變,伴表皮細胞間海綿狀水腫、表皮內水皰,有時可見角化過度或角化不全及角質層肥厚,真皮上部及血管周圍淋巴細胞和組織炎性浸潤,Loembeck等進行空腸活檢,顯示腸道嗜酸性粒細胞的超微結構有損害。
16 鑑別診斷
腸病性肢端皮炎常需與念珠菌病、大皰性表皮鬆解症和嚴重營養不良相鑑別。血漿鋅值測定有重要價值。
泛發性皮膚念珠菌病類似本病,而本病大多又繼發念珠菌感染,應予鑑別,但前者發生於肥胖或腹瀉的嬰兒,皮疹多佈於頸、腋、腹股溝等皺摺處或軀幹等部位;大皰性表皮鬆解症皮損位於易受外傷部位,水皰尼氏徵陽性;連續性肢端皮炎常先有局部外傷史,皮疹始於手指遠端,長期限於一個或幾個手指;銀屑病損害是頭部和四肢伸側紅斑鱗屑,刮屑試驗陽性;另外應與各種獲得性或條件性鋅缺乏進行鑑別。
17 腸病性肢端皮炎的治療
一般支持療法包括人乳餵養,母乳中含低分子鋅結合配體,能增加鋅的吸收;補充維生素;補充水、電解質和輸血。
二碘羥基喹啉結構與吡啶羧酸相似,能增加鋅的吸及其生物利用率,成人劑量每日200~300mg,分3次服,小兒劑量每次10~15mg/kg,一日3次,待症狀改善後逐步減量,並注意藥物副作用。
硫酸鋅內服是治療本病的首選藥物,成人0.2g,3次/d;4個月以上兒童用50mg,一日3次,4個月以下減半。治療後臨牀症狀可迅速而完全緩解,首先精神和食慾改善,其次腹瀉得到控制,皮損1周左右可消退,3~4周治癒,2個月後停藥。應根據病情及緩解程度逐步減量,並及時停藥,防止過量長期應用而出現毒副作用。葡萄糖酸酸鋅、檸檬酸鋅亦可服用。局部按亞急性皮炎對症處理。