3 概述
克魯宗綜合徵(Crouzon Syndrome)是一組由多發性顱部骨縫和麪部骨縫早閉引起的顱部和麪部複合畸形的症候羣,常伴顱內壓增高症。1921年Crouzon首先報道了此病,故稱Crouzon綜合徵,又稱鸚鵡頭綜合徵、Virchow綜合徵、先天性尖頭並指(趾)畸形綜合徵、狹顱綜合徵等,是以顱骨縫閉合過早、上頜發育不良以及眼球突出等爲主要特徵的一種綜合徵。畸形主要表現爲上頜骨嚴重後縮、突眼和反 。其發生原因可能爲多顱面縫早閉,蝶骨發育不全,導致前顱底狹窄變短、眶上緣發育不足;加之雙側冠狀縫早閉,並伸展到側顱縫和顱頂縫,造成上頜骨嚴重發育不足以及中顱凹突入眼眶的代償性膨出,眶腔過淺,不能容納整個眼球,最終產生眼球突出。該綜合徵爲常染色體顯性遺傳,國內報道在14例Crouzon綜合徵中發現4例有兩代家族史,1例有3代家族史。
。其發生原因可能爲多顱面縫早閉,蝶骨發育不全,導致前顱底狹窄變短、眶上緣發育不足;加之雙側冠狀縫早閉,並伸展到側顱縫和顱頂縫,造成上頜骨嚴重發育不足以及中顱凹突入眼眶的代償性膨出,眶腔過淺,不能容納整個眼球,最終產生眼球突出。該綜合徵爲常染色體顯性遺傳,國內報道在14例Crouzon綜合徵中發現4例有兩代家族史,1例有3代家族史。
6 別名
Crouzon綜合徵;鸚鵡頭綜合徵;先天性尖頭並指(趾)畸形綜合徵;狹顱綜合徵;Virchow綜合徵
10 克魯宗綜合徵的病因
克魯宗綜合徵大多爲常染色體顯性遺傳,也有散發者。有學者認爲,引起顱縫過早閉合和其他骨合併異常的原因是芽胚漿的變異引起兩骨的骨化中心異常接合或異常分離所致;Crouzon認爲是因胎兒期骨縫區炎症導致了早閉。
11 克魯宗綜合徵的臨牀表現
克魯宗綜合徵臨牀多見於男性。患者可出現頭顱異常、口腔頜面部異常以及眼部異常,甚至合併其他骨異常等:①頭顱異常:由於頭顱縫尤其冠狀縫的過早閉合,限制了顱骨和上頜骨等的生長,引起尖頭畸形、短頭畸形或三角形頭畸形,代償性地使額區向前明顯高起隆突,同時骨縫的骨性結合不能適應迅速發育的顱腦體積的增大,而出現腦積水、智力低下,重者顱內壓升高,頭痛,嘔吐,甚至癲癇發作;②口腔頜面部異常:主要爲上頜骨發育不良,下頜骨相對前突,而使上下頜呈反狀態,鼻部突出呈鸚鵡嘴樣,齶弓呈V形增高變尖,有時合併齶裂、脣裂,整個面中部後縮,上頜牙列擁擠,上脣短,下脣下垂,有時不出現下頜前突,反而相對形成下頜發育不全,出現強制性口呼吸等;③眼、耳異常:視力進行性下降,眼窩淺,眼球突出,淺藍色鞏膜,雙眼間距增寬、外斜視,眼裂下斜,眼球震顫,同時因視神經受壓,出現視神經炎或視盤水腫,上方視野缺如,繼發性視神經萎縮、甚至失明等,也可伴有其他眼疾,如白內障、青光眼、晶狀體異位、虹膜缺如等;雙側外耳道閉鎖,聽力減退或消失;④其他:有的可出現脊柱隱裂、並指(趾)、先天性心臟病等。
15 克魯宗綜合徵的診斷
克魯宗綜合徵的臨牀特徵主要爲突眼、上頜骨嚴重後縮呈凹盤狀臉、反畸形。有些病例如伴發顱縫早閉症可出現輕度或中度的眶距增寬症,但常爲面中部的後傾所掩蓋。其他伴發畸形可有高拱齶蓋、鼻咽腔狹小致口呼吸、睡眠打鼾及慢性間歇性呼吸困難等。部分患者可有外耳道狹窄或閉鎖,甚至有聽力障礙。由於突眼畸形,眼瞼閉合不全,可致暴露性角膜炎、角膜白斑甚或失明。Stricker(1990)按畸形的嚴重程度將克魯宗綜合徵分爲上頜型、假性型、顏面型、顱型、顱面型等5種形式,其中以顱面型畸形最爲嚴重。X線頭顱攝片,常有明顯的顱骨指壓切跡,提示存在慢性顱內壓增高。頭顱X線影定位測量對疾病的診斷和手術方案的設計有重要的參考價值。
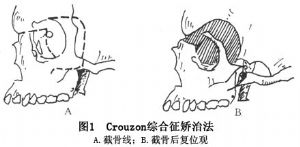
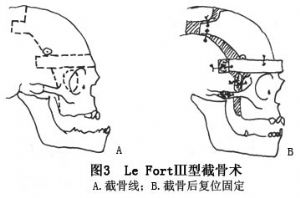
16 克魯宗綜合徵的治療
小兒克魯宗綜合徵患者可僅行額眶前移術以改善頭顱畸形和增加眶上壁的深度。輕度畸形可在青春期後進行手術治療,可選用顱外法自身穩定型的Le FortⅢ型截骨前移術(Tessier法)(圖1,2)。中、重度患者可選用顱內外聯合進路的分塊顱面骨前移及Le FortⅢ型截骨術(Enbloc手術)(Coverse、Tessier,1979)和整塊顱面骨前移及Le FortⅢ型截骨術(Monobloc手術)(Ortiz-Monasterio,1978)(圖3)。小兒伴眶距增寬症的患者,Wolfe、Tessier(1993)等建議可同期進行面中部劈開和整塊顱面骨前移及Le FortⅢ型截骨手術。