1 拼音
yī yòng fēn zǐ shāi zhì yǎng shè bèi chǎn pǐn zhù cè jì shù shěn chá zhǐ dǎo yuán zé
《醫用分子篩製氧設備產品註冊技術審查指導原則》由國家食品藥品監督管理局於2012年5月10日食藥監辦械函[2012]210號印發。
本指導原則旨在指導和規範醫用分子篩製氧設備的技術審評工作,幫助審評人員理解和掌握該類產品原理/機理、結構、性能、預期用途等內容,把握技術審評工作基本要求和尺度,對產品安全性、有效性作出系統評價。
本指導原則所確定的核心內容是在目前的科技認識水平和現有產品技術基礎上形成的,因此,審評人員應注意其適宜性,密切關注適用標準及相關技術的最新進展,考慮產品的更新和變化。
本指導原則不作爲法規強制執行,不包括行政審批要求。但是,審評人員需密切關注相關法規的變化,以確認申報產品是否符合法規要求。
3 二、技術審查要點
3.1 (一)產品名稱的要求
醫用分子篩製氧設備的命名應採用《醫療器械分類目錄》或國家標準、行業標準上的通用名稱,或以產品結構和應用範圍爲依據命名,例如**醫用分子篩製氧設備、**醫用分子篩製氧機。
3.2 (二)產品的結構和組成
製氧設備一般應包括製氧主機、流量計和溼化器。

圖1 製氧設備
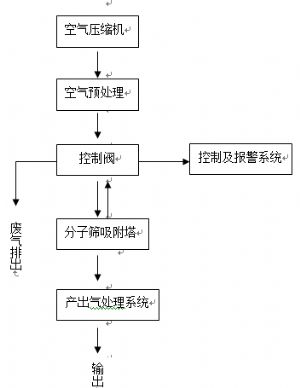
圖2 製氧設備結構組成
1.空氣壓縮機
2.空氣預處理
3.控制閥
控制經過空氣預處理系統處理的壓縮空氣,進入分子篩吸附塔,進行週期性的加壓、排氣。
在密閉的容器中,密實地填充分子篩。利用分子篩對氣體的選擇性吸附特性,分離出空氣中的氧氣。
注意:應提供氧濃度指示器,當氧濃度低於82%(V/V)時,發出報警。
6.產出氣處理系統
主要指對製氧設備產生的氧氣進行收集、過濾、調壓、溼化等處理的部分。
3.3 (三)產品工作原理
利用分子篩變壓吸附原理。工作時,向一個裝有分子篩的密閉容器內注入空氣,容器內的壓力會隨之升高,其中的分子篩隨着環境壓力的升高,大量吸附空氣中的氮氣,而空氣中的氧氣則仍然以氣體形式存在,並經一定的管道被收集起來。這個過程通常被稱爲“吸附”過程。當容器內的分子篩吸附氮氣達到一定程度時,對容器進行排氣減壓,分子篩隨着環境壓力的減小,吸附氮氣的能力下降,氮氣自分子篩內部被釋放,作爲廢氣排出。這個過程通常被稱爲“解吸”。一般的製氧設備,爲保證氧氣持續穩定的產出,多采用兩個(或多個)分子篩容器,通過控制,使一個容器處於吸附過程的同時,另一個容器處於解吸過程,反之亦然。
3.4 (四)產品作用機理
因該產品用途只是製取不小於90%(V/V)的氧氣,故本指導原則不包含產品作用機理的內容。
3.5 (五)產品適用的相關標準
目前與醫用分子篩製氧設備相關的國際標準、國家標準及行業標準列舉如下:
GB 191-2008 | 包裝儲運圖示標誌 |
GB 3096-2008 | |
GB 5832.2-2008 | |
GB 8982-2009 | 醫用及航空呼吸用氧 |
GB/T 8984-2008 | |
GB 9706.1-2007 | 醫用電氣設備 第一部分:安全通用要求 |
GB/T 14710-2009 | |
GB/T 16886.1-2011 | |
GB/T 16886.5-2003 | |
GB/T 16886.10-2005 | |
YY/T 0316-2008 | |
YY 0505-2005 | 醫用電氣設備 第1-2部分安全通用要求-並列標準電磁兼容 要求和試驗 |
YY/T 0298-1998 | 醫用分子篩製氧設備通用技術規範 |
YY 0709-2009 | |
YY 0732-2009 | 醫用氧氣濃縮器 安全要求 |
上述標準包括了註冊產品標準中經常涉及到的部件標準和方法標準。有的企業還會根據產品的特點引用一些行業外的標準和一些較爲特殊的標準。因YY 0732-2009《醫用氧氣濃縮器 安全要求》爲安全通用要求,各生產企業在註冊過程中應制定本企業的註冊產品標準。
產品適用及引用標準的審查可以分兩步來進行。首先對引用標準的齊全性和適宜性進行審查,也就是在編寫註冊產品標準時與產品相關的國家標準、行業標準是否進行了引用,以及引用是否準確。可以通過對註冊產品標準中“規範性引用文件”是否引用了相關標準,以及所引用的標準是否適宜來進行審查。此時,應注意標準編號、標準名稱是否完整規範,年代號是否有效。其次對引用標準的採納情況進行審查。即所引用的標準中的條款要求,是否在註冊產品標準中進行了實質性的條款引用。這種引用通常採用兩種方式,文字表述繁多內容複雜的可以直接引用標準及條文號。
注意“規範性應用文件”和編制說明的區別,通常不宜直接引用或全面引用的標準不納入規範性引用文件,而僅僅以參考文件在編制說明中出現。
如有新版強制性國家標準、行業標準發佈實施,產品性能指標等要求應執行最新版本的國家標準、行業標準。
3.6 (六)產品的預期用途
以空氣爲原料,利用分子篩變壓吸附工藝生產氧濃度範圍爲90%~96%(V/V)的氧氣(簡稱93%氧氣)。
3.7 (七)產品的主要風險
醫用分子篩製氧設備的風險管理報告應符合YY/T 0316《醫療器械 風險管理對醫療器械的應用》的有關要求,審查要點包括:
3.風險可接收準則,降低風險的措施及採取措施後風險的可接收程度,是否有新的風險產生。
以下依據YY/T 0316從各方面列舉了產品的危害因素,提示審查人員從以下方面考慮。
可能產生的危害 | 形成因素 |
保護接地阻抗、漏電流、電介質強度不符合要求; 應用部分與帶電部分隔離不夠; 設備的電源插頭剩餘電壓過高; 設備沒有足夠的外殼機械強度和剛度。 | |
熱能 | |
機械危險 | |
壓力 | 設備壓力超出規定值。 |
設備消音系統或運動部件損壞。 | |
配套用吸氧管、面罩生物學評價不合格。 | |
運行偏離預定的環境條件 | 有可能造成局部環境溫度升高。 |
醫用氣體的供應 | 93%氧氣的純度不符合標準要求; 93%氧氣的壓力不符合標準要求 |
不適當的標記 | 外部和內部標記不全面、標記不正確或不能夠清楚易認,以及標記不能夠永久貼牢。 |
不適當的操作說明 | 對配套用溼化杯、吸氧管等附件的使用缺少必要的警告說明和詳細的使用方法。 缺少詳細的日常使用維護規範。 |
由不熟練/未經培訓的人員使用 | 由於使用人員操作不熟練、使用不當。 |
對副作用的警告 | |
不正確的指示 | 氧氣濃度顯示或報警不準確。 |
不適當、不合適或過於複雜的使用者接口(人/機交流) | |
複雜的控制系統 | 控制系統過於複雜,使用操作時失誤。 |
維護規範缺少或不適當 | |
對醫療器械壽命的終止缺少適當的決定 | 對設備的使用壽命或終止使用的條件沒有明確規定。 |
3.8 (八)產品的主要技術指標
對產品的主要性能指標的審查,可以通過對檢驗報告內容的審查來評價是否達到了要求,檢驗報告的內容是否齊全又可以通過對產品標準的內容是否齊全來進行審查。因此產品標準的審查是產品主要技術性能指標審查中最重要的環節之一。
標準中規定的要求部分是否齊全,可以通過對是否具有以下主要內容來進行審評:
1.工作條件
是否有溫度、相對溼度、大氣壓力的要求以及電源電壓、頻率、功率等方面的要求(GB 9706.1)。
2.理化指標:
(1)氧濃度:≥90 %(V/V);
(2)水分含量:≤0.07g/m3;
(3)二氧化碳含量: ≤0.01%(V/V);
(4)一氧化碳含量:符合GB 8982-2009中表1的規定;
(5)氣態酸和鹼含量: 符合GB 8982-2009中表1的規定;
(6)臭氧及其他氣態氧化物含量: 符合GB 8982-2009中表1的規定;
(7)應無氣味;
(8)固體物質粒徑:≤10μm;
(9)固體物質含量:≤0.5mg/m3;
3.氣密性
所有氣路連接件應牢靠,不得漏氣;
4.噪聲
製氧設備的噪聲不大於60dB(A);
5.氧產量
製氧設備開機30分鐘其氧產量應達到設計要求,以L/min爲單位;
6.吸氧面罩、吸氧管(如有)
(1)如爲自制產品:
a)應有材料的要求;
c)應按照GB/T16886系列標準進行生物學評價;
(2)如爲外購產品,應採購取得醫療器械產品註冊證的產品,並且明確所使用吸氧面罩、吸氧管的規格。
7.安全性能
應符合GB9706.1、YY0732和YY0709的要求。
8.環境試驗
應符合GB/T 14710的要求。
3.9 (九)產品的檢測要求
出廠檢驗項目應至少包括外觀、氧產量、氧濃度、漏電流、電介質強度、保護接地阻抗、氣密性及控制功能驗證。
3.10 (十)產品的臨牀要求
根據《關於印發豁免提交臨牀試驗資料的第二類醫療器械目錄(試行)的通知》(國食藥監械[2011]475號)的要求,申報該產品可以提交其與已上市同類產品的對比說明,豁免提交臨牀試驗資料。
3.11 (十一)該類產品的不良事件歷史記錄
暫未見相關報道。
3.12 (十二)產品介紹、標籤和包裝標識
產品介紹一般包括使用說明書和技術介紹,兩者可合併。介紹、標籤和包裝標識應符合《醫療器械介紹、標籤和包裝標識管理辦法》的規定,並參照GB9706.1《醫用電氣設備 第一部分:安全通用要求》等標準的要求進行編寫。至少還應關注以下內容:
2. 個人、家庭使用93%氧氣時應遵從專業醫生指導的說明;
3.應有使用時遠離火源、易燃物品等警示性說明;
4.應有關於氧濃度監控、報警的說明;
5.應對產品使用方法、主要組件的壽命(分子篩更換週期)等情況做出說明;
6.不應含有誤導使用者進行吸氧的語句。
3.13 (十三)註冊單元劃分的原則和實例
原則上以結構作爲劃分註冊單元的依據,若結構、原理、性能指標等完全一致,僅產氧量不同,可視爲同一註冊單元。
舉例:產氧量爲1L/min的醫用分子篩製氧設備和產氧量爲5L/min的醫用分子篩製氧設備爲同一註冊單元。
3.14 (十四)同一註冊單元中典型產品的確定原則
典型產品應是同一註冊單元內能夠代表本單元內其他產品安全性和有效性的產品,應考慮功能最齊全、結構最複雜、風險最高的產品。
對於安全結構相同或相近的,一般情況下,較爲複雜的可以替代簡單的。註冊單元內各種產品的主要安全指標、性能指標不能被某一產品全部涵蓋時,則應選擇涵蓋安全指標、性能指標最多的產品作爲典型產品,同時還應考慮其它產品中未被典型產品所涵蓋的安全指標及性能指標。
舉例:
4 三、審查關注點
(一)產品電氣安全性能和主要技術性能指標是否執行了國家和行業的強制性標準,是否引用了適用的推薦性標準;
(二)產品的主要風險是否已經列舉,並通過風險控制措施使產品的風險在合理可接受的水平之內;
(三)介紹是否符合《醫療器械介紹、標籤和包裝標識管理規定》及相關國家、行業標準的規定。必須告知用戶的信息是否完整。
5 醫用分子篩製氧設備產品註冊技術審查指導原則編制說明
5.1 一、指導原則編寫的總體思路
本指導原則用於指導和規範個人使用的小型醫用分子篩製氧設備在註冊申報過程中審查人員對註冊材料的技術審評。
本指導原則旨在讓初次接觸該類產品的註冊審查人員對產品原理、結構、主要性能、預期用途等各個方面有個基本瞭解,同時讓技術審查人員在產品註冊技術審評時把握基本的尺度,以確保產品的安全、有效。
5.2 二、指導原則編寫的依據
《醫療器械註冊管理辦法》(局令第16號)
《醫療器械臨牀試驗規定》(局令第5號)
《醫療器械介紹、標籤和包裝標識管理規定》(局令第10號)
《醫療器械標準管理辦法》(局令第31號)
關於印發《境內第一類醫療器械註冊審批操作規範(試行)》和《境內第二類醫療器械註冊審批操作規範(試行)》的通知(國食藥監械[2005]73號)
國家食品藥品監督管理局發佈的其他規範性文件。
YY/T 0316-2008《醫療器械 風險管理對醫療器械的應用》
5.3 三、指導原則中部分具體內容的編寫考慮
(一)本指導原則僅適用於個人使用的小型醫用分子篩製氧設備。
(二)產品應適用的相關標準中給出了現行有效的國家標準、行業標準(包括產品標準、基礎標準),以及相應的國際標準。
(三)產品的主要風險中,參照YY/T0316,逐項考慮產品自身使用時可能的危害,以及技術審查時的要點。對於存在的危害,企業應在設計、驗證、檢測和改進的過程中根據風險採取有效的控制措施。
(四)產品的主要性能指標中給出了產品需要考慮的各個方面,有些需參照相關的國家標準、行業標準,有些則需要依據企業的技術能力。
(五)產品的不良事件歷史記錄主要從國家食品藥品監督管理局的不良事件數據庫中查找,也徵詢了相關領域的臨牀專家,尚未發現不良事件。
5.4 四、指導原則編寫人員
本指導原則的編寫成員由山東省醫療器械註冊技術審評人員、山東省食品藥品監督管理局行政審批人員、山東省醫療器械產品質量監督檢驗中心專家、相關生產企業專家共同組成,以充分利用各方面的信息和資源,綜合考慮指導原則中各個方面的內容,儘量確保指導原則的正確、全面、實用。