1 拼音
huà zhuāng pǐn zhōng yán liào chéng 5(CI 12075)děng 5zhǒng jìn yòng zhe sè jì de
《化妝品中顏料橙5等5種禁用着色劑的》由國家食品藥品監督管理局於2012年1月18日國食藥監保化[2012]13號發佈。
2 1 適用範圍
本方法規定了脣膏、散粉和指甲油類化妝品中酸性黃36(CI 13065)、顏料橙5(CI 12075)、顏料紅53:1(CI 15585:1)、蘇丹紅Ⅱ(CI 12140)和蘇丹紅Ⅳ(CI 26105)的高效液相色譜測定方法。
3 2 方法提要
樣品經溶劑提取後,用高效液相色譜儀分離,二極管陣列檢測器檢測,以保留時間和紫外-可見光譜圖定性,峯面積外標法定量。必要時,採用液相色譜-質譜聯用法進行確證。本方法對酸性黃36、顏料橙5、顏料紅53:1、蘇丹紅Ⅱ和蘇丹紅Ⅳ的檢出限、定量下限、檢出濃度和最低定量濃度見表1。
4 3 試劑和材料
3.1 乙腈,色譜純。
3.2 甲醇,色譜純。
3.3 四氫呋喃,色譜純。
3.4 二甲基亞碸。
3.5 乙醇,色譜純。
3.6 四丁基氫氧化銨,濃度爲55%。
3.7 檸檬酸。
3.8 氨水,濃度爲25~28%。
3.9 酸性黃36,純度≥99%。
3.10 蘇丹紅Ⅳ,純度≥94%。
3.11 蘇丹紅Ⅱ,純度≥90%。
3.12 顏料橙5,純度≥97%。
3.13 顏料紅53:1,純度≥95%。
3.14 酸性黃36標準儲備溶液( =500 μg/mL):稱取酸性黃36(3.9)對照品50 mg(精確至0.1 mg),置於100 mL容量瓶中,用甲醇(3.2)溶解並稀釋至刻度,搖勻,即得500 μg/ mL的標準儲備溶液。
3.15 蘇丹紅Ⅳ標準儲備溶液( =500 μg/mL):稱取蘇丹紅Ⅳ(3.10)對照品50 mg(按實際含量折算,精確至0.1 mg),置於100 mL容量瓶中,用四氫呋喃(3.3)和乙腈(3.1)混合液(體積比 1:9)溶解並稀釋至刻度,搖勻,即得500 μg/ mL的標準儲備溶液。
3.16 蘇丹紅Ⅱ標準儲備溶液( =500 μg/mL):稱取蘇丹紅Ⅱ(3.11)對照品50 mg(按實際含量折算,精確至0.1 mg),置於100 mL容量瓶中,用乙腈(3.1)溶解並稀釋至刻度,搖勻,即得500 μg/ mL的標準儲備溶液。
3.17 顏料橙5標準儲備溶液( =250 μg/mL):稱取顏料橙5(3.12)對照品25 mg(按實際含量折算,精確至0.1 mg),置於100 mL容量瓶中,用四氫呋喃(3.3)、二甲基亞碸(3.4)和乙腈(3.1)的混合溶液(體積比5:1:4)溶解並稀釋至刻度,搖勻,即得250 μg/ mL的標準儲備溶液。
3.18 顏料紅53:1標準儲備溶液( =250 μg/mL):稱取顏料紅53:1(3.13)對照品25 mg(按實際含量折算,精確至0.1 mg),置於100 mL容量瓶中,用二甲基亞碸(3.4)和乙醇(3.5)的混合溶液(體積比2:3)溶解並稀釋至刻度,搖勻,即得250 μg/ mL的標準儲備溶液。
3.19 着色劑混合標準儲備溶液( =50 μg/mL):分別稱取酸性黃36標準儲備液(3.14)、蘇丹紅Ⅳ標準儲備溶液(3.15)和蘇丹紅Ⅱ標準儲備溶液(3.16)各5 mL,取顏料橙5標準儲備溶液(3.17)和顏料紅53:1標準儲備溶液各10mL,置於50 mL容量瓶中,用乙腈(3.1)溶解並稀釋至刻度,搖勻,配製成質量濃度各爲50 μg/mL的標準溶液。
3.20 5種禁用着色劑系列標準溶液:精密量取一定體積的混合標準儲備溶液(3.20),置10 mL容量瓶中,加乙腈(3.1)稀釋至刻度,使得着色劑各成分的濃度分別爲0.2 μg/ mL、0.5 μg/ mL、2.0 μg/ mL、10.0 μg/mL、50.0 μg/ mL。
5 4 儀器
4.1 高效液相色譜儀:配二極管陣列檢測器。
4.2 液相色譜-串聯四極杆質譜聯用儀。
4.3 分析天平:感量爲0.0001 g。
4.4 分析天平:感量爲0.00001 g。
4.5 精密移液器。
4.6 渦旋振盪器。
4.8 高速離心機:轉速不小於10000 rmp。
6 5 分析步驟
5.1 樣品預處理
脣膏類:稱取0.5 g(精確至0.001 g)樣品,置10 mL具塞比色管中,加四氫呋喃(3.3)與乙腈(3.1)混合液(體積比1:9)至刻度,搖勻,渦旋振盪使試樣與提取溶劑充分混勻,超聲提取30 min,放至室溫,必要時,轉移至10mL具塞塑料離心管中,以10000 rpm離心5 min,上清液經0.45 μm濾膜過濾,濾液作爲試樣溶液備用。
散粉類:稱取0.5 g(精確至0.001 g)樣品,置10 mL具塞比色管中,加四氫呋喃(3.3)、二甲基亞碸(3.4)和乙腈(3.1)混合液(體積比1:1:8)至刻度,混勻,超聲提取30 min,放至室溫,上清液經0.45μm濾膜過濾,濾液作爲試樣溶液備用。
指甲油類:稱取0.5 g(精確至0.001 g)樣品,置10 mL具塞比色管中,加5~6 mL乙腈(3.1),混勻,超聲提取30min,放至室溫,加乙腈(3.1)稀釋至刻度,搖勻,靜置,必要時,轉移至10 mL具塞塑料離心管中,以10000 rpm離心5 min,上清液經0.45 μm濾膜過濾,濾液作爲試樣溶液備用。
若樣品中被測成分的濃度超過了線性範圍的上限,需對試樣溶液進行適當稀釋處理。
5.2 高效液相色譜參考條件
色譜柱:C18柱(4.6 mm×250 mm,5 μm)。
流動相:A:乙腈
B:緩衝溶液(10 mmol/L四丁基氫氧化胺(3.6),10 mmol/L 檸檬酸(3.7),用氨水(3.8)調節至pH 8.2)
時間(min) | 流動相A(%) | 流動相B(%) | 流速(mL/min) |
0.00 | 30 | 70 | 1.0 |
5.00 | 80 | 20 | 1.0 |
10.00 | 100 | 0 | 1.0 |
15.00 | 100 | 0 | 1.0 |
20.00 | 30 | 70 | 1.0 |
22.00 | 30 | 70 | 1.0 |
流速:1.0 mL/min。
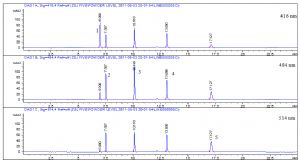
檢測波長:416 nm(酸性黃36);484 nm(顏料橙5、顏料紅53:1、蘇丹紅Ⅱ);514 nm(蘇丹紅Ⅳ)。
柱溫:30℃。
進樣量:10 μL。
5.3 樣品測定
按設定的色譜條件(5.2),取着色劑系列標準溶液(3.20)分別進樣,進行高效液相色譜分析,以系列標準溶液質量濃度爲橫座標,峯面積爲縱座標,進行線性迴歸,建立標準曲線,得到迴歸方程。取試樣溶液(5.1)進樣10 μL分析,色譜圖檢出的成分,經與該成分的保留時間和紫外-可見光譜圖比較確證後,根據測定成分的峯面積,代入迴歸方程計算各着色劑的濃度。按“6”項下計算公式計算化妝品樣品中着色劑的含量。
5.4 平行試驗
按以上步驟操作,對同一樣品獨立進行測定,獲得的兩次獨立測試結果的絕對差值不得超過算數平均值的10%。
5.5 陽性結果確證
測定過程中若有陽性結果,可採用液相色譜-質譜聯用法進一步確證。
在相同的實驗條件下,如果檢出的色譜峯的保留時間和紫外-可見光譜圖與對照品一致,所選擇的監測離子對的相對丰度比與相當濃度標準溶液的離子對相對丰度比的偏差不超過表2規定範圍,則可判定樣品中存在對應的待測成分。
相對離子丰度 | >50% | >20%至50% | >10%至20% | ≤10% |
允許的相對偏差 | ±20% | ±25% | ±30% | ±50% |
液相色譜-質譜聯用法參考條件:
色譜參考條件:
色譜柱:C18(2.1 mm×150 mm, 3.5 μm);
流動相:乙腈-0.1%甲酸水溶液系統梯度洗脫,梯度洗脫條件見表3;
柱溫:30 ℃;
流速:0.3 mL/min;
進樣量:5 μL。
表3 梯度洗脫條件
質譜參考條件:
毛細管電壓:3.5 kV;
二級錐孔電壓:5 V;
射頻透鏡電壓:0.5V;
離子源溫度:150 ℃;
脫溶劑氣溫度:500 ℃;
脫溶劑氣流量:1000L/hr;
錐孔氣流量:50 L/hr;
碰撞室壓力:3.2×10-3mbar;
低質量端1分辨率:13.0;
高質量端1分辨率:13.0;
碰撞室入口電壓:-2 eV;
碰撞室出口電壓:1 eV;
低質量端2分辨率:13.0;
高質量端2分辨率:13.0;
光電倍增器電壓:650V;
碰撞氣體:氬氣;