1 拼音
huà xué yào wù zá zhì yán jiū de jì shù zhǐ dǎo yuán zé
《化學藥物雜質研究的技術指導原則》由國家食品藥品監督管理局於2005年3月18日國食藥監注[2005]106號發佈。
指導原則編號:【H】GPH3-1
2 一、概述
任何影響藥物純度的物質統稱爲雜質。雜質的研究是藥品研發的一項重要內容。它包括選擇合適的分析方法,準確地分辨與測定雜質的含量並綜合藥學、毒理及臨牀研究的結果確定雜質的合理限度。這一研究貫穿於藥品研發的整個過程。由於藥品在臨牀使用中產生的不良反應除了與藥品本身的藥理活性有關外,有時與藥品中存在的雜質也有很大關係。例如,青黴素等抗生素中的多聚物等高分子雜質是引起過敏的主要原因。所以規範地進行雜質的研究,並將其控制在一個安全、合理的限度範圍之內,將直接關係到上市藥品的質量及安全性。
本指導原則是在借鑑國外相關指導原則[1][2]的基礎上,結合我國新藥研
發的實際情況制定的。目的是爲我國的藥品研發提供有益的指導,從而提高藥品的質量,保證人民的用藥安全。由於新藥研究的探索性很強,每種藥品的具體研究情況差異有可能很大,本指導原則不可能涵蓋雜質研究的全部,僅提供了一個基本的研究思路和方法。特殊情況下,研究單位可在科學、合理的基礎上,對雜質進行研究,只要能用科學的數據證明藥品中存在的雜質可被控制在安全、合理的範圍內,就達到了雜質研究的目的。本指導原則涵蓋的範圍包括新的及仿製已有國家標準的化學原料藥及製劑。發酵工藝生產的抗生素類藥物一般不包括在本原則的討論範疇,但如有可能,也建議參考本原則的有關要求。由於我國對臨牀研究也實行行政審批的管理,所以,本指導原則不僅適用於上述藥品的上市生產申請,也適用於臨牀研究的申請。
3 二、雜質的分類
藥品中的雜質按其理化性質一般分爲三類:有機雜質、無機雜質及殘留溶劑。按照其來源,雜質可以分爲工藝雜質(包括合成中未反應完全的反應物及試劑、中間體、副產物等)、降解產物、從反應物及試劑中混入的雜質等。按照其毒性分類,雜質又可分爲毒性雜質和普通雜質等。雜質還可按其化學結構分類,如其它甾體、其它生物鹼、幾何異構體、光學異構體和聚合物等。
本指導原則主要按照雜質的理化性質分類。
有機雜質包括工藝中引入的雜質和降解產物等,可能是已知的或未知的、揮發性的或不揮發性的。由於這類雜質的化學結構一般與活性成分類似或具淵源關係,故通常又可稱之爲有關物質。
無機雜質是指在原料藥及製劑生產或傳遞過程中產生的雜質,這些雜質通常是已知的,主要包括:反應試劑、配位體、催化劑、重金屬、其它殘留的金屬、無機鹽、助濾劑、活性炭等。
殘留溶劑是指在原料藥及製劑生產過程中使用的有機溶劑,其研究可參考有機溶劑殘留量研究的技術指導原則。
對映異構體雜質屬於雜質範疇,有關此類雜質的研究將在手性化合物研究指導原則中另行規定,本指導原則不作重複討論。 生產過程中引入的外來污染物、原料藥的不同晶型不屬於本文討論範疇。
4 三、分析方法
分析方法的選擇直接關係到雜質測定結果的專屬性與準確性,因此,在進行雜質研究時首要問題是選擇合適的雜質分析方法。
4.1 (一)分析方法的選擇
有機雜質的檢測方法包括化學法、光譜法、色譜法等,因藥物結構及降解產物的不同採用不同的檢測方法。通過合適的分析技術將不同結構的雜質進行分離、檢測,從而達到對雜質的有效控制。隨着分離、檢測技術的發展與更新,高效、快速的分離技術與靈敏、穩定、準確、適用的檢測手段相結合,幾乎所有的有機雜質均能在合適的條件下得到很好的分離與檢測。在質量標準中,目前普遍採用的雜質檢測方法主要爲高效液相色譜法(High Performance Liquid Chromatography;HPLC)、薄層色譜法(Thin Layer Chromatography;TLC)、氣相色譜法(Gas Chromatography;GC)和毛細管電泳法(Capillary Electrophoresis;CE)。應根據藥物及雜質的理化性質、化學結構、雜質的控制要求等確定適宜的檢測方法。由於各種分析方法均具有一定的侷限性,因此在進行雜質分析時,應注意不同原理的分析方法間的相互補充與驗證,如 HPLC 與 TLC 及 HPLC 與 CE 的互相補充,反相 HPLC系統與正相 HPLC 系統的相互補充,HPLC 不同檢測器檢測結果的相互補充等。
無機雜質的產生主要與生產工藝過程有關。由於許多無機雜質直接影響藥品的穩定性,並可反映生產工藝本身的情況,瞭解藥品中無機雜質的情況對評價藥品生產工藝的狀況有重要意義。對於無機雜質,各國藥典都收載了經典、簡便而又行之有效的檢測方法。對於成熟生產工藝的仿製,可根據實際情況,採用藥典收載的方法進行質量考察及控制。對於採用新生產工藝生產的新藥,鼓勵採用離子色譜法及電感耦合等離子發射光譜-質譜(ICP-MS)等分析技術,對產品中可能存在的各類無機雜質進行定性、定量分析,以便對其生產工藝進行合理評價,併爲制定合理的質量標準提供依據。
通常情況下,不揮發性無機雜質採用熾灼殘渣法進行檢測。某些金屬陽離子雜質(銀、鉛、汞、銅、鎘、鉍、銻、錫、砷、鋅、鈷與鎳等)籠統地用重金屬限度檢查法進行控制。因在藥品生產中遇到鉛的機會較多,且鉛易積蓄中毒,故作爲重金屬的代表,以鉛的限量表示重金屬限度。如需對某種(些)特定金屬離子或上述方法不能檢測到的金屬離子作限度要求,可採用專屬性較強的原子吸收分光光度法或具有一定專屬性的經典比色法(如採用藥典已收載的鐵鹽、銨鹽、硒等的檢查法檢測藥品中微量鐵鹽、銨鹽和硒等雜質)。雖然重金屬檢查法可同時檢測砷,但因其毒性大,且易帶入產品中,故需採用靈敏度高、專屬性強的砷鹽檢查法進行專項考察和控制,各國藥典收載的方法已歷經多年驗證,行之有效,應加以引用。由於硫酸根離子、氯離子、硫離子等多來源於生產中所用的乾燥劑、催化劑或pH調節劑等,考察其在產品中的殘留量,可反映產品純度,故應採用藥典中的經典方法進行檢測。如生產中用到劇毒物(如氰化物等),須採用藥典方法檢測可能引入產品中的痕量殘留物。
對於藥典尚未收載的無機雜質(如磷酸鹽、亞磷酸鹽、鋁離子、鉻離4子等)的檢測,可根據其理化特性,採用具有一定專屬性、靈敏度等的方法,如離子色譜法、原子吸收分光光度法、比色法等。
4.2 (二)分析方法的驗證
雜質檢測方法的驗證應參照相關的技術指導原則進行,重點在於專屬性和靈敏度的驗證。專屬性係指在其它成分可能共存的情況下,採用的方法能準確測定出被測雜質的特性。檢測限是反映分析方法靈敏度的一個重要指標,所用分析方法的檢測限一定要符合質量標準中對雜質限度的要求,最低檢測限不得大於該雜質的報告限度。
爲驗證雜質分析方法的專屬性,對於原料藥,可根據其合成工藝,採用各步反應的中間體(尤其是後幾步反應的中間體)、立體異構體、粗品、重結晶母液等作爲測試品進行系統適用性研究,考察產品中各雜質峯及主成分峯相互間的分離度是否符合要求,從而驗證方法對工藝雜質的分離能力。
爲了考察方法能否有效檢測出原料藥或製劑中的降解產物,還可根據藥物的化學結構特點、製劑的處方與工藝、儲存條件等選用合適的酸、鹼、光、熱、氧化反應等加速破壞性試驗來驗證分析方法的專屬性,必要時可採用二極管陣列檢測器、質譜檢測器等檢測峯的純度。因爲在強制降解試驗條件下產生的降解產物較藥品貨架期產生的降解產物複雜、未知雜質多,分離難度大,上述分析方法可有效地顯示各色譜峯的純度,以免因分離度不符合要求,導致分析結果的不準確。如不具備檢測峯純度的試驗條件,可通過適當調整流動相的組成或比例使各色譜峯的相對保留時間發生改變,用同一份經加速破壞試驗的供試品溶液進樣,然後比較流動相調整前後雜質峯的個數;也可採用TLC法比較同一份經加速破壞試驗的供試品溶液在不同展開系統下的斑點個數及位置,以此佐證雜質分析方法的專屬性。 強制降解試驗對於未知雜質的分離度考察是非常必要的,其目的主要是提供關於雜質(特別是降解物)與主成分的分離情況、樣品穩定性及降解途徑等重要信息。在試驗過程中,應注意破壞性試驗要適度,應着重考察敏感條件。如產品在一定條件下穩定,則無必要再提高條件的劇烈程度進行重複試驗。破壞試驗的程度暫無統一要求,一般以強力破壞後主成分的含量仍佔絕大部分爲宜。此時已產生了一定量的降解產物,與樣品長期放置的降解情況相似,考察此情況下的分離度更具有實際意義。要達到這種破壞程度,需要在研究過程中進行摸索,先通過初步試驗瞭解樣品對光、熱、溼、酸、鹼、氧化條件的基本穩定情況,然後進一步調整破壞性試驗條件(如光照強度、酸鹼濃度、破壞的時間、溫度等),以得到能充分反映降解產物與主成分分離的結果和圖譜。另外,通過比較試驗前後主峯面積的變化,還可粗略估算降解物對主成分的相對響應因子,瞭解樣品在各種條件下的穩定性,爲包裝及貯藏條件的選擇等提供信息。對於性質相對穩定的藥品,如有充分的文獻依據或試驗數據,則可以免做強制降解試驗。
4.3 (三)有機雜質的定量方法
有機雜質的檢測一般多采用HPLC法,有時也採用TLC、GC等其它方法。如採用HPLC法,須採用峯面積法,具體定量方法有①外標法(雜質對照品法)、②加校正因子的主成分自身對照法、③不加校正因子的主成分自身對照法、④峯面積歸一化法。①法定量比較準確,採用時應對對照品進行評估和確認,並制訂質量要求。②法應對校正因子進行嚴格測定,僅適用於已知雜質的控制。③法的前提是假定雜質與主成分的響應因子基本相同。一般情況下,如雜質與主成分的分子結構相似,其響應因子差別不會太大。④法簡便快捷,但因各雜質與主成分響應因子不一定相同、雜質量與主成分量不一定在同一線性範圍內、儀器對微量雜質和常量主成分的積分精度及準確度不相同等因素,所以在質量標準中採用有一定的侷限性。 有關物質中包括已知雜質和未知雜質。已知雜質對主成分的相對響應因子在0.9-1.1範圍內[3]時,可以用主成分的自身對照法計算含量,超出0.9-1.1範圍時,宜用雜質對照品法計算含量,也可用加校正因子的主成分自身對照法。理想的定量方法爲已知雜質對照品法與未知雜質不加校正因子的主成分自身對照法兩者的結合。研究人員可根據實際情況選用合適的定量方法。 在選擇合適的分析方法時還應考慮生產能力及質量控制的可行性等技術因素。儘管在附件中規定的限度精確到小數點後第二位,但並不意味着在日常的生產質控中所用的分析方法也要如此精確。如經過必要的驗證,也可採用薄層色譜法等分析方法。在研發過程中,如果分析方法有改變,則應進行方法改變前後所得分析結果的可比性研究。
對於 TLC 法,通常採用雜質對照品法和主成分自身對照法進行檢控制,後者僅限於雜質斑點的顏色與主成分斑點顏色一致的情況下使用。
5 四、雜質檢測數據的積累
雜質檢測數據的積累是制訂質量標準中雜質限度的重要依據之一,它包括藥品研製過程中所有批次樣品(包括用於安全性、臨牀研究的樣品)的雜質檢測數據。應該對大於報告限度的各雜質的檢測結果進行彙總,各雜質應以編號或保留時間作爲標識以便區分識別。
檢測結果應提供具體試驗數據(如雜質的保留時間及含量),不能籠統地表述爲“符合要求”或“合格”等。每批樣品中大於報告限度的任何雜質都應在其檢測報告中加以體現和說明,如要放寬附件 1 及 2 中雜質的報告限度,則應提供合理的依據。大於報告限度的任何雜質均應統計在內,並計入總雜質中。如雜質含量小於 1.0%,則報告的數據應精確到小數點後第二位;如雜質含量大於 1.0%,則報告的數據可精確到小數點後第一位。建議採用表格的形式,列出每批樣品的批號、批量、生產日期與地點、生產工藝、單個雜質及總雜質的含量、產品的用途(如臨牀研究、穩定性考察等)與所用分析方法有關的參考文獻。對於製劑,還應註明所用原料藥的批號、製劑的內包裝及其封閉物及貯存條件等。
方法學研究中雜質分離度和檢測限的圖譜、代表性批次的圖譜、採用其它雜質檢測方法所得的圖譜、加速及長期穩定性試驗的圖譜等,可以輔助說明產品中雜質的概況。如有必要,申報單位還應提供所有批次產品的雜質概況(如色譜圖等)。
建議列表說明每一次安全性研究與臨牀研究用樣品的原料藥的批號。 藥物研發者應將藥品在合成、純化、製劑製備與貯存過程中實際或可能產生的雜質儘量全面地加以總結,還應對合成過程中引入的雜質、可能會由原材料帶入成品中的雜質、降解產物、原料藥與輔料或內包裝材料、封閉物之間的反應產物等做出評估。對合成過程中引入雜質的評估,應僅限於對現有化學反應條件下可能產生的雜質。對檢測雜質所做的研究工作,包括小試與中試樣品的雜質實測結果、以及爲了鑑定樣品貯存過程中可能產生雜質而進行的加速破壞降解試驗的結果等,均應進行歸納總結,從而爲雜質限度的確定提供參考。此外,還應對整個研發過程中的實驗室規模、中試規模樣品的雜質情況進行比較,如果雜質的種類、數量及含量不一致,則應進行合理的分析。
對於超過鑑定限度的雜質應作進一步的研究,確定其來源,推測其可能的結構,進而判斷該雜質對藥物安全性的影響;對於在穩定性研究中產生的超過鑑定限度的降解產物也應做相應的研究。對於未超過鑑定限度的雜質一般不需進行結構研究。對於可能具有特殊的生理活性或毒性的雜質,則應進行結構確證和安全性驗證。
在雜質研究時,應根據具體的生產工藝,對原料藥製備過程中涉及到的無機物進行檢測,根據整個研發過程中的實驗室規模、中試規模樣品的實測情況,對催化劑、重金屬等無機雜質帶入成品中的可能性進行評估,就質量標準中是否收載這些無機雜質檢測項目進行必要的討論說明,並提供相關的試驗數據和文獻依據。
6 五、雜質限度的制訂
質量標準中應詳細說明各雜質的檢測方法及其限度。在制訂質量標準中雜質的限度時,首先應從安全性方面進行考慮,尤其對於有藥理活性或毒性的雜質;其次應考慮生產的可行性及批與批之間的正常波動;還要考慮藥品本身的穩定性。在質量標準的制訂過程中應充分論證質量標準中是否收載某一雜質檢測項目及其限度制訂的合理性。可根據穩定性考察、原料藥的製備工藝、製劑工藝、降解途徑等的研究及批次檢測結果來預測正式生產時產品的雜質概況。當雜質有特殊的藥理活性或毒性時,分析方法的定量限及檢出限應與該雜質的控制限度相適應。設定的雜質限度不能高於安全性數據所能支持的水平,同時也要與生產的可行性及分析能力相一致。在確保產品安全的前提下,雜質限度的確定主要基於中試規模以上產品的實測情況,考慮到實際生產情況的誤差及產品的穩定性,往往對限度做適當放寬。如果各批次間的雜質含量相差很大,則應以生產工藝穩定後的產品爲依據,確定雜質限度。
除降解產物和毒性雜質外,已在原料藥質量標準中控制,且在製劑過程中含量沒有增加的雜質,製劑中一般不再控制。
6.1 (一)有機雜質的限度確定
質量標準中對有機雜質的限度規定應包括:每一個已知雜質、未知雜質及總雜質。共存的異構體和抗生素的多組分一般不作爲雜質進行控制,必要時作爲共存物質在質量標準中規定其比例。單一的對映體藥物,其對映異構體應作爲雜質控制。
由於創新藥物與仿製藥情況不同,在確定雜質限度時,可有所區別,所以本指導原則在此分別予以說明。
1、創新藥物
創新藥物是指國內外均未上市的新的化學實體及其製劑。由於在創新藥物的研究過程中,需通過一系列的藥理毒理及臨牀研究來驗證該藥品的安全有效性,而研究所用的樣品本身會包含一定種類與數量的雜質,所以如果在這些研究中並未明顯反映出與雜質有關的毒副作用,即使有些雜質的含量超出了附件 1 或 2 的質控限度,仍可認爲該雜質的含量已經通過了安全性的驗證。在此前提之下,如果該雜質的含量同時也在正常的製備工藝所允許的限度範圍內,那麼根據試驗樣品中雜質的含量所確定的限度可認爲是合理的。由於動物與人在毒性反應上的差異、臨牀試驗例數的限制,致使在新藥申請上市時的安全性數據仍很有限,據此制定的雜質限度尚不能完全保證產品的安全性,故新產品應在上市後繼續監測不良反應,並對新增不良反應的原因進行分析。如與雜質有關,則應分析原因,設法降低雜質含量,這樣制訂出來的雜質限度才能保證產品的安全性。如某雜質同時也是該藥物在動物或人體中的主要代謝產物,則對該雜質可不考慮其安全性,但需制訂合理的限度。
對於用於某些適應症的藥物,可以根據用藥人羣、劑量、用藥週期、臨牀經驗、利弊權衡等,對雜質的限度做適當的調整。當研究證明某些藥物中的雜質與不良反應有關,則應在制訂該雜質的限度時引起重視,並適當提高限度要求。反之,雜質的限度可適當放寬。由此可見,在特殊情況下,應具體問題具體分析,在保證安全的前提下,可以修改附件 1 或 2 中的限度,並同時提供修改限度的充分理由。
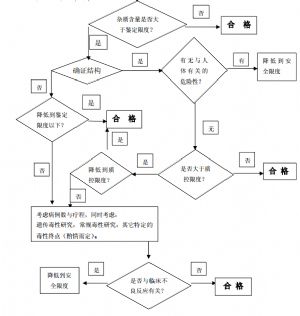
當雜質的限度大於附件 1 或 2 中的規定時,可根據附件 3 中的決策樹來考慮下一步的研究。在某些情況下,將雜質的限度降到符合附件 1 或 2的要求,可能比提供該雜質的安全性數據更爲簡單。如果能有比較充足的文獻數據證明該雜質的安全性,也可不降低該雜質的限度。如果以上兩種途徑均不可行,則應考慮進行必要的安全性研究,其結果的可靠性與一系列因素有關,如病例數、日劑量、給藥途徑與療程等。儘管直接用分離純化的雜質進行安全性研究比較合適,但也可採用含有雜質的原料藥進行研究。
2、仿製已有國家標準的藥品
對於仿製已有國家標準的藥品,可以根據已有的標準制訂相應的雜質限度。如果該標準中未規定雜質的限度,應與已上市同品種藥品(建議首選原研發企業在有效期內的產品)進行全面的質量對比研究,分析其雜質的種類與含量,根據研究的結果,以及穩定性考察的結果,決定是否需在質量標準中對雜質進行控制。如果難以獲得已上市同品種的標準,但有相同原料藥的其它劑型上市,則在制訂雜質限度時,可參考此上市產品質量標準,對雜質進行控制。
由於工藝或處方的不同導致在研產品與已上市同品種產品的雜質種類不同,仿製產品中新雜質的含量高於附件 1 或 2 規定的合理限度,或在研產品的雜質含量明顯高於已上市的同品種產品的雜質實測值。爲了保證產品的安全性,應考慮優化產品的處方與製備工藝,將雜質的含量降到規定的質控限度以內。如仍不能達到要求,則應做必要的安全性研究。
3、其它新藥
改變給藥途徑的製劑,其雜質限度的確定參照創新藥物的要求進行。 對於其它類別的新藥,如果能夠獲得已上市的對照樣品,則可按照仿製已有標準的藥品的研究思路,在詳細的質量對比研究的基礎上,確定雜質的限度。如果不能獲得對照樣品,則應參照創新藥物的要求確定雜質限度,或通過詳細的安全性試驗來證明已有的雜質限度是安全的。
6.2 (二)無機雜質的限度確定
無機雜質的限度主要根據該雜質的毒性、對藥品本身質量(如穩定性)的影響及各批次產品的實測結果而定。如果某些產品的無機雜質在放置過程中會增加,則制訂該雜質的限度時,還應綜合考慮穩定性考察的結果。
各國藥典收載的質量標準及我國已批准上市產品的註冊標準中包含有各類無機雜質的限度,在這些限度以內的無機雜質可以認爲其安全性已得到了確認。因此,這些限度對於我們確定在研產品的無機雜質限度具有重要的參考價值。要注意根據在研產品的給藥途徑、適應症、劑量等選擇合適的參考標準,確定合理的限度。
7 六、臨牀研究申請與上市生產申請階段的雜質研究[4][5]
我國對藥品的註冊審批分爲臨牀研究與上市生產兩個階段。在申報臨牀研究時,雜質研究工作可從以下幾方面考慮。1.爲了保證臨牀研究受試者的安全,在申報臨牀研究前,應對已有批次產品的雜質進行比較全面的檢測,根據安全性研究用樣品的雜質含量情況來證明臨牀研究用藥品是安全的。2.由於藥品的研發過程是一個不斷完善的過程,隨着研究的深入,可能會對雜質的分析方法做相應的改進。所以,在雜質含量初步得到控制的前提下,可在臨牀研究期間對雜質分析方法進行完善。3.對於創新藥物,雜質限度的最終確定需根據臨牀研究結果進行綜合權衡。故在申報創新藥物臨牀研究時,可對雜質的限度做一個初步的規定。
臨牀研究結束後,應將放大生產的樣品與臨牀研究樣品中的雜質進行詳細比較,如因生產規模放大而產生了新的雜質,或已有雜質的含量超出原有的限度時,同樣應根據附件 1 或 2 來判斷該雜質的含量是否合理,如不合理,則應參照決策樹來考慮下一步的研究工作。
8 七、結語
雜質的研究是藥品研究的重要方面,它貫穿於整個藥品研究的始終。
藥品中的雜質是否能得到合理、有效的控制,直接關係到藥品的質量可控性與安全性。在進行雜質研究時應重點關注以下幾個方面:1.應注意對雜質檢測方法的選擇與驗證。2.應注意對研究過程中所有批次的樣品,包括各種生產規模的樣品中的雜質進行完整的記錄,這些數據將是制訂雜質限度的一個重要依據。3.應特別注意,在確定雜質的限度時,一定要綜合考慮雜質的安全性、生產的可行性與產品的穩定性。在確定仿製藥品的雜質限度時,應與已上市產品進行質量對比研究,以確保產品的安全性。
9 八、名詞解釋
報告限度(Reporting Threshold):超出此限度的雜質均應在檢測報告中報告,並應報告具體的檢測數據。
鑑定限度(Identification Threshold):超出此限度的雜質均應進行定性分析,確定其化學結構。
質控限度(Qualification Threshold):質量標準中一般允許的雜質限度,如制訂的限度高於此限度,則應有充分的依據。


11 十、參考文獻
1 . ICH Harmonized Tripartite Guideline: Impurities In New Drug Substances,Q3A(R),7 Feb,2002.
2 . ICH Harmonized Tripartite Guideline: Impurities In New Drug Products,Q3B(R),5 Feb,2003.
3.中華人民共和國藥典 2005 年版附錄增修訂內容彙編,國家藥典委員會。
4. INDs for Phase 2 and Phase 3 Studies Chemistry, Manufacturing, and Controls Information
5. Content and Format of Investigational New Drug Applications (INDs) for Phase 1 Studies of Drugs, Including Well-Characterized, Therapeutic,Biotechnology-derived Products
12 十一、著者
《化學藥物雜質研究的技術指導原則》課題研究組