1 拼音
xuè zhàn jì shù cāo zuò guī chéng (2012bǎn )
《血站技術操作規程(2012版)》由衛生部於2011年12月31日衛醫政發〔2012〕1號印發,自2012年6月1日起施行,《中國輸血技術操作規程(血站部分)》(衛醫發〔1997〕第28號)同時廢止。
2 前言
《中國輸血技術操作規程(血站部分》自1997年發佈以來,對促進血站規範化管理起到重要的作用。隨着輸血科學技術的進步和血液管理工作要求的提高,原有內容已經不適應當前需要,爲此衛生部組織專家重新編制《血站技術操作規程(2012版)》(以下簡稱《規程》)。
本《規程》正文包括獻血者健康檢查、全血採集、血液成分製備、血液檢測、血液隔離與放行和質量控制6個部分,對所涉及的關鍵技術要點做出相應規定。其中一些原則性的規定,血站在制定自身的操作規程時應當根據實際情況進一步細化。以“宜”表述的內容爲推薦性內容。
本《規程》的附錄爲資料性附錄,供血站參考。
各血站應當按照國家相關法律、法規、規章、技術規範和標準,以及本《規程》的要求,結合具體工作實際,編制適合本血站使用的技術操作規程。
本《規程》自2012年6月1日起施行。
3 1 獻血者健康檢查
1.1 目的
按照GB18467《獻血者健康檢查要求》(以下簡稱《要求》)的規定,對具有獻血意向的潛在獻血者進行健康檢查,對檢查結果進行綜合分析和判斷,做出是否適合獻血的結論,保障獻血者健康和安全。
1.2 覈對獻血者身份
將獻血者本人相貌與其有效身份證件原件覈對。有效身份證件包括居民身份證、居民社會保障卡、駕駛證、軍(警)官證、士兵證、港澳通行證和臺胞證以及外國公民護照等。
覈查獻血者身份無誤後,將獻血者身份信息錄入血液管理信息系統(以下簡稱BMIS),具體錄入方式有:1)用身份證識讀器讀取身份信息並存入BMIS;2)在《獻血登記表》手寫登記,隨後手工錄入BMIS,注意覈對信息填寫和輸入的正確性。
1.4 詢問和查詢既往獻血史
詢問獻血者和查詢BMIS有無既往獻血史。如獻血者曾獻血,獻血間隔期應符合要求,不處於被暫時或永久屏蔽狀態。
1.5 履行告知義務
1.6 健康徵詢
請獻血者仔細閱讀、理解並如實回答獻血前健康徵詢問題(見《要求》),體檢人員給予必要的指導和溝通。
1.7 知情同意
請獻血者簽名,表明獻血者已正確理解獻血前須知內容並如實回答獻血前健康徵詢問題,自主、自由地決定是否獻血。
1.8 一般檢查
按照《要求》規定,對獻血者進行一般檢查,常規項目包括體重、血壓、脈搏等,必要時測量體溫。具體檢查方法見衛生部《臨牀護理實踐指南》。記錄健康檢查結果和結論並簽名。
在獻血前採集獻血者血液標本做血液檢測。在採集血液標本前應覈對獻血者身份。檢測項目包括血紅蛋白(Hb)和丙氨酸氨基轉移酶(ALT)檢測。血紅蛋白檢測可採用目測法,如硫酸銅目測法(見附錄A)或試紙條比色法,必要時進一步用儀器檢測。ALT採用幹化學法/速率法記錄檢測結果和結論並簽名。
1.10 健康檢查結論
1.10.1 將獻血者健康徵詢、一般檢查以及血液檢測的結果與《要求》的規定進行對照分析和評價,做出獻血者是否符合獻血條件的判斷並簽名。
1.10.2 將健康檢查結果和結論與獻血者溝通。對於需要永久屏蔽獻血的,做好解釋工作;對於暫時不適宜獻血的,告知不適宜獻血的情形解除後,經體檢合格可以獻血。
4 2 全血採集
2.1 獻血場所配置
獻血場所的人員、設施、設備和器具、關鍵物料的配備按有關規定執行。
2.2 採血人員準備
2.2.1 心理調適
採血人員調整好心理與情緒,進入獻血者服務工作狀態,情緒穩定,工作熱情,說話和氣,態度和藹,耐心細緻。
2.2.2 技術準備
熟悉採血技術操作規程,尤其應注意關鍵控制點和近期變更的操作步驟。
2.2.3 着裝與配飾 採血人員着工作制服,不佩帶戒指、手鐲(鏈)等飾物。
2.2.4 手衛生 採血人員保持手衛生,具體操作按照《WS/T313 醫務人員手衛生規範》的規定執行。
2.3 採血器材準備
2.3.1 採血器材清單 建立採血器材卡片,列出採血位所需的全部器材。採血人員按卡片準備和核查採血器材的種類和數量。採血器材的數量與預計採血量相適宜。一次性使用物品在有效期內且包裝完好。採血器材準備工作應有專人複覈。
2.3.2 血袋 1)無破損、無滲漏,無污染,抗凝劑和保養液無變色;2)處於有效期內;3)宜採用具有留樣袋的血袋。
2.3.3 標本管 1)帶有分離膠用於檢測病毒核酸的標本管;2)用於酶聯免疫吸附法(以下簡稱ELISA)、ALT和血型檢測的標本管。
2.3.4 消毒劑 1)一般選用含碘消毒劑,對碘過敏者可選用其他消毒劑;2)所用消毒劑應當符合相應的國家標準要求;3)處於有效期內;4)標明啓用日期。
2.3.5 採血儀(秤) 開啓並檢查採血儀(秤),證實正常運行。
2.3.6 熱合機 開啓並檢查熱合機,證實處於正常狀態。
2.4 獻血者身份覈對
2.5.1 在血液採集過程中應當加強與獻血者的溝通,尤其是進行每一項主要操作之前,應當與獻血者溝通並取得配合。
2.5.2 詢問獻血者的既往獻血經歷、近日休息等情況,評估出現獻血不良反應的可能性和不適合獻血的情況。
2.5.3 觀察獻血者面部表情和肢體語言,是否處於緊張、害怕甚至恐懼狀態。如發現這些不利情況,則不急於採血,做好寬慰工作,待獻血者解除思想顧慮,充分放鬆後開始準備採血。
2.6.1 穿刺部位的選擇 應選擇無損傷、炎症、皮疹、皮癬、疤痕的皮膚區域爲穿刺部位。
2.6.2 穿刺靜脈的選擇 1)選擇上肢肘部清晰可見、粗大、充盈飽滿、彈性好、較固定、不易滑動的靜脈;2)常選擇的靜脈主要有肘正中靜脈、頭靜脈、前臂正中靜脈、貴要靜脈等;3)用食指指腹上下左右觸摸,確定其位置、粗細和彈性,評估並確定穿刺位點和路徑;4)使用止血帶可使靜脈充盈,便於觸及和穿刺。
2.7.1 用無菌棉拭蘸取適量使用濃度消毒劑,以穿刺點爲中心,自內向外螺旋式旋轉塗拭,消毒面積不小於6 cm×8 cm。作用1~3min 。宜消毒2~3遍。
2.7.2 不應觸摸已消毒的皮膚,不應靠近已消毒的皮膚講話。
2.8.2 採取相應措施(如用止流夾夾住血袋導管)防止空氣進入血袋。手持針柄,取下護針帽,按照預先選定的穿刺部位進行穿刺。
2.8.3 穿刺路徑爲自皮膚穿刺點進入,皮下組織前行約0.5~1.0 cm,進入靜脈腔,前行約0.5~1.0 cm。
2.8.4 如需第二次穿刺,應當在徵得獻血者同意後,在另一手臂選擇穿刺部位和靜脈,使用新採血袋的採血針進行穿刺。
2.9 血液採集和混勻
2.9.1 靜脈穿刺成功後,如果使用的帶留樣袋的採血袋,鬆開留樣袋夾子,使最先流出的血液流入留樣袋,約15~20 ml,用做血液檢測標本。夾閉留樣袋夾子,鬆開阻塞件下端止流夾,使血液流入採血袋。如果使用不帶留樣袋的採血袋,鬆開夾子,使血液直接流入採血袋。
2.9.2 固定針頭位置,用敷料保護穿刺點。
2.9.3 維持靜脈穿刺點與血袋的落差,保持血流通暢。囑獻血者做握拳和鬆手動作,以促進靜脈迴流。血流不暢時,及時調整針頭位置。當不易觀察血流時,應注意觀察穿刺部位有無異常及血袋重量是否遞增。
2.9.4 血液開始流入採血袋後,即將其與抗凝劑輕勻混合。宜採用連續混合採血儀。如果採用手工混合,應當至少每90秒混合1次,充分混勻。
2.9.5 應當對採血時間進行控制。200 ml全血採集時間>5 min,或400 ml全血採集時間>10 min,應給予特殊標識,所採集的全血不可用於製備血小板。200ml全血採集時間>7 min,或400 ml全血採集時間>13 min,所採集的全血不可用於製備新鮮冰凍血漿。
2.9.6 與獻血者進行交流,觀察獻血者面容、表情,及時發現並處置獻血反應。
2.10.1 採血量達到要求時,囑獻血者松拳,鬆開止血帶,合閉止流夾,用創可貼/消毒棉球/紗布輕按靜脈穿刺點,撥出針頭後即加重按壓,用彈力繃帶包紮,鬆緊度適中。
2.10.2 囑獻血者在獻血者休息處用茶點,休息10~15 min。
2.11.1 應當印製獻血後注意事項,並將其發給每位獻血者。
2.11.2 獻血後注意事項主要有:1)穿刺點上的敷料應保留至少4小時;2)多補充水分,食用易消化的食物和水果,避免飲酒,保證充足的睡眠;3)獻血後24小時內不劇烈運動、高空作業和過度疲勞;4)工作人員的聯繫方式,如果存在獻血前沒有如實告知的可能影響血液安全的高危行爲,或者獻血後感覺明顯不適或異常,請其及時聯繫工作人員。
2.12 致謝
2.13 留取標本
2.13.1 檢測結果用於判定血液能否放行的標本只能在獻血時同步留取,不得在獻血者健康檢查時提前留取。
2.13.2 如果使用帶留樣袋的採血袋,將留樣針插入真空採血管,留取血樣。
2.13.3 如果使用不帶留樣袋的採血袋,將靜脈穿刺針插入真空採血管,留取血樣。應單手操作,避免手被針頭刺傷。
2.14.1 一次只能對來源於同一獻血者的一份血袋、標本管和獻血記錄進行標識。經覈對後,將惟一性條形碼標識牢固粘貼在採血袋、標本管、轉移袋、血袋導管、獻血記錄單上。
2.14.2 宜在標本管與留樣針/靜脈穿刺針分離前開始標識,對採血袋和標本管的標識應當首先連續完成,不應中斷。
2.14.3 宜在標本管與留樣針/靜脈穿刺針分離前核查採血袋、血液標本、獻血登記表,所標識的獻血條形碼應一致。宜採用計算機程序進行覈查。
2.15 熱合
2.15.1 分段熱合血袋導管,以供交叉配血、血型複查和血液標本保存使用。血袋應保留注滿全血的導管至少20 cm。
2.15.2 在熱合過程中不應用力牽拉或扭轉導管,待焊極鬆開1~2秒後方可取出已封口的導管。
2.15.3 應檢查熱合部位,如有滲漏,則重新熱合,並評估對血液無菌性的影響。
2.15.4 熱合分離針頭,將其放置在利器盒內。
2.17.2 血液標本採集後應儘快處理,在合適的溫度下保存。
2.18 獻血現場整理
2.18.2 盤點採集血液、標本、獻血登記表數量,應當一一對應,保證準確無誤。
2.18.4 盤點物料消耗。
5 3 血液成分製備
3.1.1 血液成分品種見GB18469《全血及成分血質量要求》。
3.2 製備環境
3.2.3 用於製備血液成分的開放系統,製備室環境應達到10000級、操作檯局部應達到100級(或在超淨臺中進行)。
3.2.4 製備需要冷藏的血液成分時,應儘可能縮短室溫下的製備時間。
3.3 設備
3.3.1 設備數量及功能應能滿足製備工作的要求。
3.3.2 應建立和實施設備的確認、維護、校準和持續監控等管理制度,實施惟一性標識及使用狀態標識,以確保設備符合預期使用要求。
3.4 物料
3.4.2 物料質量及其生產和供應方的資質應符合相關法規的要求。
3.4.3 物料使用前,應檢查有效期、外觀質量等,確認符合質量要求後方可使用。對不合格物料應進行標識、隔離,防止誤用。
3.4.4 製備方法 製備新的血液品種或製備條件發生明顯改變時,應對血液製備方法進行確認。
3.5 起始血液
3.5.1 用於製備血液成分的起始血液應符合GB18469《全血及成分血質量要求》的要求。
3.5.2 起始血液的保存和運輸應當符合國家有關規定的要求。
3.5.3 接收起始血液時,應覈對數量,檢查外觀、血袋標籤等內容,確認符合質量要求後方可用於血液成分製備。
3.6 製備方法
3.6.1 離心
3.6.1.1 根據所製備血液成分要求和離心機操作手冊,確定離心轉速、加速和減速、離心時間和溫度等參數,編制離心程序。
3.6.1.4 離心程序應經過確認,應能分離出符合質量要求的血液成分。
3.6.1.5 對已經投入常規使用的離心程序的變更實施控制,定期檢查覈對,防止被非授權修改。
3.6.1.6 每批血液製備的離心記錄應包括離心操作者簽名和所採用的離心程序。
3.6.2 分離
3.6.2.1 離心結束後,從離心機中取出離心杯,從離心杯中取出血袋,避免振動,進行目視檢查,觀察離心效果、血袋及其導管有無滲漏,離心杯中有無血跡,如有破損應查找滲漏點。血袋破漏的,應作消毒和報廢處理。
3.6.2.2 將血袋置於分漿夾或血液分離機。將不同分層的血液成分轉移至密閉系統的轉移聯袋中,以最大限度收集目的成分(紅細胞、血小板、血漿等),並且使不需要的其他成分的殘留量最小的方式進行分離和轉移。
3.6.3 速凍
3.6.3.1 速凍是保存凝血因子Ⅷ的關鍵加工步驟,冷凍速率和血漿中心溫度是2個關鍵參數。
3.6.3.2 應當使用專用設備,按操作介紹進行冷凍操作。
3.6.3.3 應當將擬速凍的血袋逐袋平放,而不應重疊堆放。
3.6.3.4 應當將新鮮冰凍血漿和冷沉澱凝血因子快速凍結,最好在60 min內將血漿中心溫度降至-30℃以下。
3.7 標識
3.7.1 使用聯袋製備時,在原袋和轉移袋分離之前,應當檢查每個血袋上獻血條碼的一致性。宜採用計算機系統進行覈對,以避免人爲差錯。
3.7.2 需要連接新的血袋(過濾、分裝等)時,應當保證每一血袋獻血條碼一致。宜採用按需打印方式產生標籤,粘貼完畢,經計算機系統覈對無誤後,纔給予斷離。
3.7.3 應當對血液製備過程中發現的疑似不符合品進行標識和隔離,以進一步調查和判斷。
3.8 目視檢查
3.8.1 在接收、離心、分離、熱合及交付的各個環節應對每袋血液進行目視檢查。
3.8.2 目視檢查內容主要有:是否有滲漏、標籤是否完整、血液外觀是否正常。
3.8.3 目視檢查發現異常的,應給予標識、隔離及進一步處理。
3.9 質量記錄
3.9.1 製備記錄主要有:血液交接、製備,設備使用與維護,製備環境控制,醫療廢物處理等。
3.9.2 製備記錄應可追溯到起始血液、製備人員、製備方法、製備環境、使用設備和物料。
3.9.3 製備記錄宜以電子記錄爲主,以手工紙面記錄爲補充。
3.10.1 多聯袋製備血液成分。
3.10.1.1 紅細胞和冰凍血漿的製備 1)第1次重離心後將盡可能多的血漿轉移至轉移袋;2)將紅細胞保存液袋內的紅細胞保存液轉移至紅細胞袋,充分混合即爲懸浮紅細胞;3)覈對血袋上的獻血條形碼,如一致則熱合斷離,生成1袋懸浮紅細胞和1袋血漿;4)血漿紅細胞混入量少(目視觀察)即可將血漿袋熱合斷離;5)如血漿紅細胞混入量較多,應當經過第2次重離心後,把上清血漿轉移至已移空的紅細胞保存液袋,熱合斷離(如欲製備冷沉澱,則不熱合斷離);6)將血漿速凍,低溫保存。
3.10.1.2 紅細胞、濃縮血小板和冰凍血漿的製備(富血小板血漿法):1)第1次輕離心後將富含血小板血漿轉移至轉移袋;2)將紅細胞保存液袋內的紅細胞保存液轉移至紅細胞袋;3)覈對血袋上的獻血條形碼,如一致則熱合斷離,生成1袋懸浮紅細胞和1袋富血小板血漿;4)將富含血小板血漿袋重離心,上清爲血漿,沉澱物爲血小板;5)留取適量血漿,將多餘的血漿轉移至已經移空的紅細胞保存液袋,熱合斷離,生成1袋濃縮血小板和1袋血漿;6)將血漿袋速凍,低溫保存;7)將濃縮血小板袋在室溫靜置1~2小時,待自然解聚後,輕輕均勻血袋,製成濃縮血小板混懸液,在22±2℃的環境下振盪保存。
3.10.1.3 紅細胞、濃縮血小板和冰凍血漿的製備(白膜法):1)第1次重離心後,將血漿轉移至第1個轉移袋,將適量血漿及白膜層轉移至第2個轉移袋;2)將紅細胞保存液袋內的紅細胞保存液轉移至紅細胞袋,充分混合即爲懸浮紅細胞;3)覈對血袋上的獻血條形碼,如一致則熱合斷離懸浮紅細胞袋和血漿袋;4)將白膜成分袋和1個空袋一起進行輕離心,將富含血小板血漿(上層)轉移至空袋,製成濃縮血小板,熱合斷離,棄去白細胞袋。
3.10.2.1 用於製備冷沉澱凝血因子的起始血液爲新鮮冰凍血漿。
3.10.2.2 離心法
3.10.2.2.1 取出待制備冷沉澱的新鮮冰凍血漿,置4±2℃冰箱中過夜融化或在4±2℃水浴裝置中融化。
3.10.2.2.2 當血漿基本融化時,取出血漿,在4±2℃的環境下重離心。
3.10.2.2.3 將大部分上層血漿移至空袋,製成冰凍血漿。將留下的20~30ml血漿與沉澱物混合,製成冷沉澱凝血因子。
3.10.2.3 虹吸法
3.10.2.3.1 將新鮮冰凍血漿袋置於4±2℃水浴裝置中,另一空袋懸於水浴箱外,位置低於血漿袋,兩袋之間形成一定的高度落差。
3.10.2.3.2 血漿融化後,隨時被虹吸至空袋中,當融化至剩下40~50 ml血漿與沉澱物時,閉合導管,阻斷虹吸。將血漿與沉澱物混合,製成冷沉澱凝血因子。將冰凍血漿袋和冷沉澱凝血因子袋熱合斷離。
3.10.3 洗滌紅細胞製備
3.10.3.1 待用洗滌溶液聯袋提前放置冷藏保存,無破損滲漏,溶液外觀正常,在有效期內。
3.10.3.2 將合格的紅細胞懸液用作製備洗滌紅細胞懸液的起始血液,無破損滲漏,血液外觀正常,在有效期內。
3.10.3.3 使用無菌接合機將待洗滌的紅細胞懸液袋導管和洗滌溶液聯袋進行無菌接合連通。
3.10.3.4 將洗滌溶液移至紅細胞袋內,液體量約爲100ml/單位,夾緊導管,混勻。
3.10.3.6 離心後將血袋取出,避免震盪,垂直放入分漿夾中,把上清液轉移至空袋內,夾緊導管。
3.10.3.7 重複3.10.4~3.10.6步驟,洗滌3次。
3.10.3.8 將適量(50 ml/單位)保存液(生理鹽水或紅細胞保存液)移入已完成洗滌的紅細胞,混勻。
3.10.3.9 熱合,貼籤,入庫。
3.10.3.10 如果是在開放環境製備,應嚴格遵從無菌操作。
3.10.3.11 如果在開放環境製備或最後以生理鹽水混懸,洗滌紅細胞保存期爲24小時。如果是在閉合無菌環境中製備且最後以紅細胞保存液混懸,洗滌紅細胞保存期與洗滌前的紅細胞懸液相同。
3.10.4 去除白細胞
3.10.4.1 應當使用白細胞過濾技術去除全血或紅細胞懸液中的白細胞。
3.10.4.2 根據白細胞過濾器生產方介紹的要求進行過濾操作。
3.10.4.3 應當在密閉環境(使用白細胞過濾多聯血袋或無菌接合技術)製備。
3.10.4.4 應當在採血後2天內(採血次日爲第1天)完成白細胞過濾。
3.10.4.5 檢查待濾過血液的外觀,並充分混勻後進行過濾。
3.10.4.6 如果在進行白細胞過濾操作前,血液已經處於保存溫度(4±
2℃),需要在室溫進行過濾時,室溫應≤26℃,而且應當儘快放回至既定保存溫度的環境中,從取出到放回的時間應<3小時。
3.10.4.7 如果在白細胞過濾後,將血液轉移至不屬於原聯體血袋的其他血袋,應當建立與實施標識控制機制,保證過濾後血液的正確標識。
3.10.5 冰凍紅細胞
3.10.5.1.1 取擬冰凍保存的全血或懸浮紅細胞,離心去除上清液,用無菌接合技術將紅細胞轉移至容量適當的、適宜於冰凍保存的轉移袋內。
3.10.5.1.2 在無菌條件下,緩慢滴加複方甘油溶液至紅細胞袋內,邊加邊振盪,使其充分混勻。
3.10.5.1.3 在室溫中靜置平衡30 min,置-65℃以下保存。
3.10.5.2 冰凍紅細胞的解凍 從低溫冷凍保存箱中取出冰凍紅細胞,立即放入37℃~40℃恆溫水浴箱中,輕輕振動使其快速融化,直至冰凍紅細胞完全解凍。
3.10.5.3 洗滌除去甘油 將專用洗滌鹽液袋與解凍紅細胞袋無菌接合,採取滲透壓梯度遞減方法洗滌。最後1次的洗滌上清液應無明顯溶血跡象。
3.10.5.4 使用自動化設備製備冰凍和解凍紅細胞時,按照設備使用說明書進行操作。
3.10.6.1 根據設備操作介紹設置醫用血漿病毒滅活光照櫃的參數。
3.10.6.3 用無菌導管連接設備或百級淨化臺內按無菌操作技術將血漿袋與病毒滅活血袋連接。
3.10.6.4 將血袋懸掛於支架上,打開導管夾,使血漿經“亞甲藍添加元件”,流入光照袋。
3.10.6.6 光照處理後的血漿經病毒滅活裝置配套用輸血過濾器過濾,濾除亞甲藍和絕大部分白細胞,即得病毒滅活血漿。
3.10.7 血液輻照
3.10.7.1 輻照室應符合《電離輻射防護與輻射源安全基本標準》的要求。
3.10.7.2 按照輻照儀使用說明書設置輻照參數。
3.10.7.3 血液輻照最低劑量爲25 Gy,血液任何位點的輻照劑量不宜超過50 Gy。
3.10.7.4 紅細胞在採集後14天內可輻照,輻照後可再儲存14天。
3.10.7.5 血小板在保存期內均可輻照,輻照後可保存至從採集算起的正常保存期限。
3.10.7.6 粒細胞宜在採集後儘快輻照,輻照後宜儘快輸注。
3.10.7.7 在輻照過程中應嚴格區分未輻照和已輻照血液的標識。
6 4 血液檢測
4.1.1 人類免疫缺陷病毒(HIV)感染標誌物及其檢測方法有2種選擇,可任選其中1種:1)採用2個不同生產廠家的ELISA試劑檢測HIV-1和HIV-2抗體或聯合檢測HIV-1和HIV-2抗原和抗體;2)採用1種ELISA試劑檢測HIV-1和HIV-2抗體或聯合檢測HIV-1和HIV-2抗原和抗體,採用1種試劑檢測HIV核酸。
4.1.2 乙型肝炎病毒(HBV)感染標誌物及其檢測方法有2種選擇,可任選其中1種:1)採用2個不同生產廠家的ELISA試劑檢測乙型肝炎表面抗原(HBsAg);2)採用1種ELISA試劑檢測HBsAg,採用1種試劑檢測HBV核酸。
4.1.3 丙型肝炎病毒(HCV)感染標誌物及其檢測方法有2種選擇,可任選其中1種:1)採用2個不同生產廠家的ELISA試劑檢測HCV抗體或聯合檢測HCV抗原和抗體;2)採用1種ELISA試劑檢測HCV抗體或聯合檢測HCV抗原和抗體,採用1種試劑檢測HCV核酸。
4.1.4 ALT採用2種方法(幹化學法和速率法)進行2次檢測,分別在採血前和採血後進行。
4.1.5 梅毒螺旋體感染標誌物及其檢測方法:採用2個不同生產廠家的ELISA試劑檢測梅毒特異性抗體。
4.2 血液檢測試劑
4.2.1 試劑選擇
4.2.1.3 有條件的實驗室可自行開展試劑評價(見附錄B)。不具備條件的實驗室可充分利用國家專業機構(如中國食品藥品檢定研究院、中國疾病預防控制中心、衛生部臨牀檢驗中心等)的評價數據。
4.2.2 證照要求
4.2.2.1 應建立血液檢測試劑證照審覈程序,在採購前和驗收時覈實應具備的有效證照文件。
4.2.2.2 採購藥品類檢測試劑應索取以下加蓋供貨單位印章的資料存檔:1)《藥品生產許可證》或者《藥品經營許可證》和營業執照複印件;2)《藥品生產質量管理規範》或者《藥品經營質量管理規範》認證證書複印件;3)藥品的批准證明文件複印件;3)供貨單位藥品銷售委託書;4)銷售人員有效身份證明覆印件;5)血源篩查試劑的批簽發文件;6)出廠質量檢驗報告。
4.2.2.3 採購醫療器械類檢測試劑應索取以下加蓋供貨單位印章的資料存檔:1)《醫療器械生產企業許可證》或者《醫療器械經營企業許可證》和營業執照複印件,對生產、經營屬於《醫療器械分類目錄》中第一類醫療器械的企業,只需索取營業執照複印件;2)醫療器械註冊證書和《醫療器械註冊登記表》複印件;3)供貨單位醫療器械銷售委託書;4)銷售人員有效身份證明覆印件;5)出廠質量檢驗報告。
4.2.3 進貨檢查驗收 應建立並執行進貨檢查驗收制度,檢查驗收內容主要有:1)驗明藥品合格證明和其他標識;2)外觀檢查(運輸包裝箱完整無損,運輸冷鏈符合要求,試劑包裝盒完整無損,無液體泄漏);3)到貨數量和銷售憑證(購貨單位、試劑、供貨商等名稱,規格、批號、數量、價格)
4.2.4 隔離存放 應將通過進貨檢查驗收後的試劑進行隔離存放,防止誤用。
4.2.5 質量抽檢
4.2.5.1 應建立並執行試劑的質量抽檢制度,應對每次購進的試劑進行質量抽檢。
4.2.5.2 應將試劑介紹列入文件控制範圍。應對試劑介紹版本和內容進行檢查。其操作要求如已變更,實驗室的試驗操作在試劑啓用時應同時變更,嚴格控制未按試劑介紹進行試驗操作的情形發生。
4.2.5.3 試劑盒組成、組分性狀與介紹一致,無泄漏,足量,標識正確。
4.2.5.4 用於質量抽檢的樣本有:1)試劑盒對照;2)室內質控品;3)實驗室自制或商品化的血清盤。前2種爲必須,後1種爲可選。
4.2.5.5 質量抽檢結果要求:1)試劑盒對照品檢測結果符合試劑介紹要求,2)室內質控品檢測結果符合既定要求;3)如果適用,實驗室自制或商品化的血清盤符合既定要求。
4.2.6 審覈批准
4.2.6.1 應由授權人對採購驗收和質量抽檢的過程和結果進行審覈,批准其用於血液檢測。
4.2.6.2 應建立和保存試劑採購驗收、質量抽檢和審覈批准的記錄(見附錄C)。
4.2.7 試劑保存和質量監控
4.2.7.1 應對經批准使用的試劑進行標識和放行,宜對試劑盒粘貼“可用”標籤。
4.2.7.2 應按試劑介紹要求的保存條件進行保存,應在有效期內使用。
4.2.7.3 應對試劑的庫存(批號、失效期、庫存量等)進行管理,防止試劑過期或者中斷。
4.2.7.4 在試劑保存和使用過程中應注意試劑性能出現衰減,如果試劑盒對照品和室內質控品的檢測值呈現連續走低趨勢且無法使其回升糾正,應考慮終止使用。
4.3 儀器設備使用要求
4.3.1 新的或者經過大修的檢測設備在正式投入正常使用之前應經過確認。新設備的確認應包括安裝確認、運行確認和性能確認。經過大修的設備根據需要進行適當確認,必要時應進行計量檢定或校準。
4.3.2 按照檢測設備用戶手冊要求進行操作,包括使用、校準、維護等工作。
4.3.3 如果使用多臺設備檢測同一個項目,應對設備之間的性能和差異進行比較。
4.3.4 應定期檢查自動化檢測設備試驗參數的設置,應保存檢查記錄。
4.3.5 在試驗過程中自動化檢測設備出現故障需要進行手工操作時,應注意自動化設備操作和手工操作的銜接及其對結果的影響。應記錄手工操作步驟和操作者。
4.4.1 應使用實驗室信息管理系統對整個檢測過程(從標本接收、試驗、結果和結論判定)進行信息化管理。
4.4.2 實驗室信息管理系統的功能應包括:1)標本接收;2)試驗項目選擇;3)試驗數據記錄與彙總;4)試驗數據的計算;5)試驗結果的判定;6)血液篩查結論的判定;7)血液篩查結論傳輸至血液管理信息系統併爲其所利用。
4.4.3 實驗室信息管理系統運行參數的設置應建立權限控制。應保存設置參數的書面記錄,並定期將其與實際設置參數對照,確保設置無誤,應保存覈實記錄。
4.5.1 血液標本的一般要求 1)標本與血液、獻血者一一對應;2)標本質量符合檢測項目技術要求;3)標本信息具有可追溯性。
4.5.2.1 本血站實驗室應與血液標本採集和送檢部門進行充分溝通與協商,共同制定標本採集和送檢程序,質量管理部門應予以審覈。
4.5.2.2 血液集中化檢測的委託方和受託方應進行充分溝通與協商,共同制定標本採集和送檢程序,雙方質量管理部門應予以審覈,並經雙方法定代表人批准。
4.5.2.3 標本採集和送檢程序的要點有: 1)標本類型及檢測項目、標本量、標本管、標本運輸及包裝要求;2)標本的惟一性標識(條形碼);3)標本的質量要求;4)標本採集、送檢和接收;5)標本信息和檢測報告信息的傳輸與接收,檢測報告時限;6)如爲集中化檢測,檢測的委託方和受託方的標識與聯繫方式。
4.5.3.1 應根據每項試驗的技術要求,採用相應類型的真空採血管留取檢測標本。試管應無裂痕、無滲漏,容量應滿足檢測項目要求。
4.5.3.2 將病毒核酸檢測結果列入血液放行依據的實驗室,應按照試劑介紹要求選擇合適的標本管,最好採用含惰性分離膠的EDTAK2真空採血管留取核酸檢測標本。
4.5.4.1 應對血液標本採集前的準備、標本的採集、標識、登記和保存過程實施有效控制,一次只對一袋血液和同源血液標本管貼籤,確保標本與血液、獻血者一一對應,貼籤無誤。標本質量符合檢測項目技術要求。
4.5.4.2 檢測結果用於血液放行的血液標本,應在採集血袋血液的同時或者從血袋血液中留取。
4.5.4.3 血液標本的採集與標識的具體操作見本規程第2章全血採集。
4.5.5.1 可以電子或紙面方式登記標本信息,應進行覈對,防止信息錄入錯誤。可通過網絡、傳真或其它形式傳輸標本信息。
4.5.5.2 核酸檢測標本採集後,根據所採用的採血管和試驗的技術要求實施離心。
4.5.5.3 血液標本在採血現場的臨時保存溫度爲2~10℃。
4.5.6.1 標本應隔離密封包裝,包裝材料應滿足防水、防破損、防外泄、保持溫度、易於消毒處理。裝箱時應保持標本管口向上。
4.5.6.2 對於送交集中化檢測實驗室的標本的包裝要求主要有:1)可使標本運輸在過程中保持在2~10℃;2)外包裝有明確標識(放置朝向、易碎)和交付接收雙方的聯繫方式。
4.5.6.3 標本應在2~10℃條件下運輸,應對運輸過程的冷鏈效果進行確認並定期監測。
4.5.6.4 應對標本運輸過程進行記錄,其要點有:1)啓運時間、地點;2)運抵時間、地點;3)標本箱編號、標本類型、數量;4)運輸包裝有無受損、有無泄漏;5)運輸時間2小時以上的應記錄箱內溫度;6)標本交運人、承運人;7)運輸過程中發生的可能影響標本質量的意外事件及處理措施。
4.5.7.1 接收時標本應覈查:1)標本來源、數量、採集時間;2)標本採集管使用正確與否;3)標本是否滿足既定的質量要求;4)標本與送檢單信息對應性和完整性。
4.5.7.2 如發現溢漏應立即將尚存留的標本移出,對溢出標本管和原包裝箱進行消毒並記錄,必要時報告實驗室負責人和送檢單位。
4.5.7.3 應拒收標本的情形有:1)檢測申請關鍵信息缺失或不符;2)標本管上無標識或標識不清、不正確;3)標本管選用錯誤;4)標本量不足或被稀釋;5)不符合試劑介紹要求的情形。
4.6 試驗操作
4.6.1 按照試劑生產方提供的試劑使用說明書進行操作。
4.6.2 如需對個別試驗參數進行修改,應進行確認。
4.6.3 宜採用自動化檢測設備操作標本和試劑加樣以及試驗過程。
4.6.4 自動化設備運行參數的設置應建立權限控制。應保存設置參數的書面記錄,並定期將其與實際設置參數對照,確保設置無誤,應保存覈實記錄。
4.6.5 應保存自動化檢測設備運行記錄,並定期對運行狀態進行審覈。
4.6.6 自動化檢測設備運行時,如果需要人工輔助或干預,應對實施人工輔助或干預的人員、人工輔助或干預的時間和內容、與自動化檢測設備運行的銜接等進行記錄。
4.6.7 如果是採用手工操作標本和試劑加樣或試驗微孔板,應完整記錄每一加樣和操作步驟。
4.7 試驗性能監控
4.7.1 一般要求
4.7.1.1 在血液篩查過程中,應對試驗性能持續進行監測,以發現正在發生的任何性能變化,這些變化如果沒有得到及時糾正,最終可能導致試驗批次的失敗,或者弱陽性樣本的漏檢。
4.7.1.2 選擇能夠及時反映試驗性能或試驗應用(試驗或者試驗操作者/操作系統)變化的1項或者多項參數進行監測,以保證試驗性能。這些參數包括:1)試驗對照的檢測值;2)質控品的檢測值;3)初、複試有反應率及兩者的比例(複試有反應性標本數/初次試驗有反應性標本數)。
4.7.1.3 應當根據質控品的特性和用途加以選擇和使用。
4.7.1.4 作爲試驗有效性的外部對照應具備:1)經過國際(國家)標準溯源;2)以合適的基質進行稀釋;3)檢測標誌物含量接近檢測限(S/CO值爲2~5)。
4.7.1.5 用於監測試驗穩定性的質控品,不要求經過國際(國家)標準溯源。
4.7.1.6 實驗室應當儘可能使用質控品。
4.7.1.7 質控品所含目標檢測物的濃度應滿足實驗要求。在日常使用前應對質控品的種類、規格、外觀、批號和效期進行檢查。
4.7.1.8 質控品應與血液檢測標本在相同的檢測條件下進行檢測。如單一微板爲一批試驗,每一微板至少包括1份外部質控品。
4.7.1.9 外部質控品和內部(試劑盒)對照不可相互替代。
4.7.1.10 外部質控品的用途(用於判定試驗有效性或監控試驗穩定性)一經確定,不應隨意更改。
4.7.2 ELISA室內質控(參見附錄D)。
4.8 試驗結果的判定
4.8.1 試驗結果判定規則 應制定明確的試驗有效性和標本試驗結果判定規則,將其編寫或設置成爲計算機程序,對其編寫、設置、修改和啓用應進行控制,所有修改均應保存原版本,確保其具有可追溯性。
4.8.2 試驗有效的判定
4.8.2.1 應覈查每批試驗所使用的試劑、設備、試驗過程、有無人工干預或其他非正常工作步驟出現等關鍵控制點,正確無誤後方可對試驗有效性進行判定
4.8.2.2 試劑盒各種試驗對照的檢測值符合試劑介紹的要求,是判定試驗有效的最低要求。
4.8.2.3 如果外部質控品的檢測值是作爲試驗有效性的判定依據之一,其檢測值應符合既定要求。
4.8.2.4 如果外部質控品的檢測值是作爲試驗穩定性的監控,其檢測值應符合既定範圍。如果超出既定範圍,按既定程序決定試驗是否有效。
4.8.2.5 如果採用人工判定,應詳細記錄每一判定依據,應雙人覈查。
4.8.2.6 如果判定一批試驗無效,則該批試驗所有標本的檢測結果均爲無效。
4.8.3 標本試驗結果計算和判定 判定試驗有效後,按照試劑介紹的要求計算臨界值和灰區。根據標本檢測值與臨界值的比較結果,判定爲標本檢測結論爲無反應性、有反應性或不確定。
方案1:
以同一試驗對原血樣(或從血袋導管重新取樣)做雙孔複試,如果雙孔複試結果均爲無反應性, 其初試有反應性可能由於假反應性或技術誤差導致,檢測結論爲無反應性,血液可放行供臨牀使用;如果雙孔複試結果中任何1孔爲有反應性, 則檢測結論爲有反應性,對應的血液及由其製備的所有成分應隔離並報廢,將血液標本轉送相關實驗室做進一步確證或補充試驗(圖4-1)。
方案2:
不做重複試驗,初次試驗結論即爲檢測最終結論(圖4-1)。

圖4-1 初次試驗有反應性的處理方案
4.11.1 血型檢測項目 GB18469《全血及成分血質量要求》規定的強制檢測項目有:1) ABO血型正確定型;2)RhD血型正確定型。
4.11.2 血型檢測方法 血型篩查常用方法有平板法和微板法、血型鑑定常用試管法和微板法。按照試劑介紹和《全國臨牀檢驗操作規程》(第3版)的規定進行具體操作和質量控制。附錄E提供了建立微板法一般方法,可供實驗室自行建立微板法時參考。全自動化血型鑑定系統按照使用說明書進行操作,投入使用前應經過充分確認。如有必要,可增加血型基因檢測。
4.11.3.1 應當經過2次檢測結果的比對,一致時才能做出血型檢測最終結論。
4.11.3.2 如果2次檢測結果不一致,應當進行細緻審慎調查,發現導致不一致的原因,正確無誤加以解決。
4.12 血液檢測最終結論的判定
4.12.1 血液檢測合格判定標準 HIV、HBV、HCV、梅毒感染標誌物檢測的最終結論均爲無反應性,ABO/RhD血型正確定型,ALT測定值符合國家相關標準。
4.12.2 血液檢測不合格的判定標準 不符合4.12.1條規定的情形。
4.12.3 應當建立和實施血液檢測最終結論的計算機判定程序。如果需要人工判定,應由雙人複覈。
4.13 血液檢測結論的報告和利用
4.13.1 血液檢測最終結論是血液放行與否的依據。只有檢測合格的血液方可放行供臨牀使用,檢測不合格的血液不得放行。
4.13.2 血液檢測最終結論應以電子數據傳輸,併爲計算機血液放行控制程序直接利用。
4.13.3 如果需要人工錄入血液檢測最終結論,或者需要人工放行,應由雙人複覈。
7 5 血液隔離與放行
5.1.1 待檢測、製備等尚未被判定合格的血液和不合格的血液應被物理隔離,防止不合格血液的誤發放。
5.1.2 應設立有明顯標識的合格品區、隔離區和不合格品區。
5.1.3 設施設備應衛生、整潔,並定期清潔。
5.1.5 進出血液隔離區域的血液應做好交接和記錄,記錄至少包括血型、品名、數量、時間、交接人及簽名等。
5.2 血液放行
5.2.1 經過培訓考覈的被授權人員才能承擔放行工作,質量管理人員對血液的放行進行監控,並留有監控記錄。
5.2.2.1 對檢測不合格、外觀不合格、異常採集和符合保密性棄血等的血液,應進行標識,並移入不合格品區。
5.2.2.2 將檢測報告中尚未最終判定結果的血液繼續隔離並做好標識。
5.2.3 貼籤
5.2.3.2 一次只對一袋血液貼籤。
5.2.3.3 合格血液或合格血液製備的每一種血液成分只能印製惟一的合格血液標籤。該合格血液標籤印有惟一的條形碼。
5.2.3.4 需要複製惟一的合格血液標籤,應由被授權者確認原先印製的合格血液標籤已被銷燬。
5.2.3.5 通過惟一的條形碼可以追溯到獻血者、用血醫院以及血液採集、檢測、保存、發放等全過程記錄。
5.2.3.6 合格血液標籤的內容符合《血站質量管理規範》的要求。
5.2.3.7 粘貼標籤前應檢查確認血袋無破損、無滲漏,血液外觀無異常。
5.2.3.9 粘貼合格血液標籤後應能清楚觀察血液外觀,並且不會影響血袋透氣性。
5.2.3.10 合格血液標籤粘貼於血袋後應再次確認該標籤粘貼無誤。
5.2.3.12 已經放行進入合格品庫的血液,再經製備、分裝、轉換後,應重新粘貼具有惟一性條形碼的合格血液標籤,並保證粘貼無誤和可追溯性。
5.3 計算機控制
5.3.2 需要人工放行時,應建立與實施複覈制度。
8 6 質量控制
6.1 導則
6.1.1 範圍
質量控制包括全血及血液成分質量檢查、關鍵物料質量檢查、關鍵設備質量檢查和環境衛生質量檢查。
6.1.2 抽樣數量 全血及血液成分和關鍵物料質量檢查的抽樣量爲每月製備量的1%或至少4袋。若該成分血每月製備量少於4袋的,在保證質量的前提之下,由血站自行制訂抽樣頻率和數量。
6.1.3 取樣方法 應當儘可能採用密閉系統進行取樣。如果採用開放系統取樣,應當嚴格無菌操作。取樣對血液質量沒有構成影響的,取樣後的血液可發放使用。
6.1.4 控制指標 全血及血液成分的質量控制指標遵從GB18469《全血及成分血質量要求》的規定,關鍵物料、關鍵設備和環境衛生的質量控制指標遵從有關規定。
6.1.5 檢測機構 血站可自行檢測,也可委託具備相應檢測能力的檢測機構(計量檢定機構、血站、醫院、疾病預防控中心等)進行檢測。
6.1.6 趨勢分析 應當對血液質量控制抽檢結果進行趨勢分析,出現異常趨勢時,應當組織有關部門實施調查和回顧,並採取相應的改進措施。進行趨勢分析時,應當充分考慮到獻血者個體差異,對於由於獻血者個體差異所引起的,而且在血液採集和製備過程中難以控制的不影響血液安全性的指標(見表6-1),如果有75%的抽檢結果落在質量控制指標範圍內,可認爲血液採集和製備過程受控。
6.2.1 檢查項目 不同血液品種的質量控制檢查項目見表6-1。
外觀 | 容量* | 無菌試驗 | Hb* | 遊離Hb* | 白細胞殘留量* | 血漿蛋白含量* | 上清液蛋白含量* | pH* | Ⅷ因子活性* | 纖維蛋白原含量* | 甘油殘留量* | 中性粒細胞計數* | 亞甲藍殘留量* | ||||||
√ | √ | √ | √ | √ | √ | ||||||||||||||
√ | √ | √ | √ | √ | √ | √ | |||||||||||||
√ | √ | √ | √ | √ | √ | ||||||||||||||
√ | √ | √ | √ | √ | √ | √ | |||||||||||||
√ | √ | √ | √ | √ | √ | √ | |||||||||||||
√ | √ | √ | √ | √ | √ | √ | √ | ||||||||||||
√ | √ | √ | √ | √ | √ | √ | |||||||||||||
(保存期爲24 h) | √ | √ | √ | √ | √ | ||||||||||||||
√ | √ | √ | √ | √ | √ | √ | √ | ||||||||||||
√ | √ | √ | √ | √ | √ | √ | |||||||||||||
√ | √ | √ | √ | √ | √ | √ | √ | ||||||||||||
√ | √ | √ | √ | √ | √ | √ | √ | ||||||||||||
√ | √ | √ | √ | √ | √ | ||||||||||||||
√ | √ | √ | √ | √ | √ | √ | |||||||||||||
√ | √ | √ | √ | √ | |||||||||||||||
(亞甲藍光化學法) | √ | √ | √ | √ | √ | √ | |||||||||||||
√ | √ | √ | √ | √ | √ | ||||||||||||||
√ | √ | √ | √ | √ | √ | ||||||||||||||
√ | √ | √ | √ | √ | √ | ||||||||||||||
注1:“√”爲適用檢查項目;注2:“ *”爲適用於“75%的抽檢結果落在質量控制指標範圍內,可認爲血液採集和製備過程受控”的檢查項目
6.3.1.2 質量標準
6.3.1.2.1 產品標識:塑料採血袋標記產品名稱、型式代號、採血袋(無採血袋時按轉移袋)公稱容量和國家標準編號組成。塑料採血袋分爲單袋(S),雙聯袋(D),三聯袋(T),四聯袋(Q)和轉移袋(Tr)五種型式。如符合國家標準要求,採血袋公稱容量爲400mL的雙聯袋(D)的產品標記爲:血袋D-400
6.3.1.2.2 系統密閉性:塑料採血袋的採血針、採血管、輸血插口必須連成一個完整的密閉系統,保證採集、分離、輸注和儲存血液時其內腔不與外界空氣相接觸。
6.3.1.2.3 血袋袋體外觀:塑料採血袋袋體應無色或微黃色,無明顯雜質、斑點、氣泡。塑料採血袋內外表面應平整,在滅菌過程中和在溫度不超過40℃的貯存期內不應有粘連。塑料採血袋熱合線應透明、均勻。採血管和轉移管內外表面光潔,不應有明顯條紋、扭結和扁癟。袋中的抗凝保存液及添加液應無色或微黃色、無渾濁、無雜質、無沉澱。
6.3.1.2.4 標籤應字跡清楚,項目齊全。標籤應有下列內容:
2)公稱容量(採血量);
4)“無菌”、“無熱原”限定條件的說明,“一次性使用”、“用後銷燬”字樣,使用說明,保存的血液條件;
5)注意事項:發現滲漏、長黴、混濁等變質現象,禁止使用;
6)產品名稱和標記;
7)生產廠家名稱、地址和商標;
6.3.1.3 檢查方法:在光線明亮處,以目力檢查;以擠壓方式檢查系統密閉性。
所選用的血袋必須符合國家相關標準,每一批血袋必須有出廠檢驗報告。
6.3.1.5 規格:必須符合使用要求。
6.3.2.2 質量標準
6.3.2.2.1 每個注射器的單包裝上應有下列標誌:
1)生產廠家名稱或商標;
4)一次性使用;
5)包裝如有破損禁止使用;
6)若帶注射針頭,應註明規格;
6.3.2.2.2 外觀:注射器應清潔、無微粒和異物。注射器外套必須有足夠的透明度,能毫無困難地讀出劑量,能清晰地看到基準線。
6.3.2.2.3 潤滑:注射器應有良好的潤滑性能。注射器的內表面(包括橡膠活塞),不得有明顯可見的潤滑劑匯聚。
所選用的注射器必須符合國家相關標準,每一批注射器必須有出廠檢驗報告。
6.3.2.5 規格:必須符合使用要求。
6.3.3.2 質量標準
6.3.3.2.1 外觀:去白細胞濾器外殼應光潔,無明顯機械雜質、異物,焊接面應均勻、無氣泡,軟管應柔軟、透明、光潔,無明顯機械雜質、異物、扭結。
6.3.3.2.2 每個單包裝上應有以下內容:
4)標明適用範圍的產品標記;
5)製造商和/或經銷商名稱、地址;
6)單包裝內不應有肉眼可見異物。
所選用的去白細胞濾器必須符合國家相關標準,每一批去白細胞濾器必須有出廠檢驗報告。
6.3.3.5 規格:必須符合使用要求。
6.3.4.2質量標準
6.3.4.2.1 外觀:病毒滅活輸血過濾器的軟管應光潔,無明顯機械雜質、異物、扭結。過濾部件、亞甲藍添加元件外殼應光潔,無明顯機械雜質、異物,焊接面應均勻、無氣泡。
6.3.4.2.2 每個單包裝上應有以下內容:
4)標明適用範圍的產品標記;
5)製造商和/或經銷商名稱、地址;
6)單包裝內不應有肉眼可見異物。
所選用的病毒滅活輸血過濾器必須符合國家相關標準,每一批病毒滅活輸血過濾器必須有出廠檢驗報告。
6.3.4.5 規格:必須符合使用要求。
6.3.5.2 質量標準
6.3.5.2.1 外觀:包裝完整,標識清晰。
6.3.5.2.2 每個單包裝上應有以下內容:
4)標明適用範圍的產品標記;
5)製造商和/或經銷商名稱、地址。
所選用的單採耗材必須符合國家相關標準,每一批單採耗材必須有出廠檢驗報告。
6.3.5.5 規格:必須符合使用要求
6.3.6.2 質量標準 標籤的底色應爲白色,標籤應潔淨、無破損,字跡清楚;標籤上文字一般爲實體黑色字體。
所選用的血袋標籤必須符合國家相關標準,每一批血袋標籤必須有出廠檢驗報告。
6.3.6.5 規格:必須符合使用要求。
6.3.6.6 擬採用新的供應方所提供的標籤的確認方法參見附錄G。
6.3.7.1 抽樣
6.3.7.1.2 外購成品硫酸銅溶液:每批至少隨機抽檢5套。
6.3.7.2 質量標準
用於男性獻血者血比重檢查的硫酸銅溶液比重,在20℃時應爲1.0520,允許誤差爲±0.0005。用於女性獻血者血比重檢查的硫酸銅溶液比重,在20℃時應爲1.0510,允許誤差爲±0.0005。
用於男性獻血者血比重檢查的硫酸銅溶液比重,在溶液允許的使用溫度範圍時應爲1.0520,允許誤差爲±0.0005。用於女性獻血者血比重檢查的硫酸銅溶液比重,在溶液允許的使用溫度範圍時應爲1.0510,允許誤差爲±0.0005。
6.3.7.3 檢測方法:見《中華人民共和國藥典》(2010年版)二部 附錄 “韋氏比重秤法”。
所選用的硫酸銅溶液必須符合國家相關標準,每一批外購硫酸銅溶液必須有出廠檢驗報告。
6.3.7.5 規格:必須符合使用要求。
6.3.8.2 質量標準
試管上的標誌、標籤應清晰。試管應無色透明、光滑、平整,正常視力能清楚觀察到試管內血樣;不得有明顯變形、沙眼、氣泡、雜質等。
所選用的真空採血管必須符合國家相關標準,每一批真空採血管必須有出廠檢驗報告。
6.3.8.5 規格:必須符合使用要求。
6.3.9.1 檢驗試劑包括:乙型肝炎病毒表面抗原診斷試劑盒、丙型肝炎病毒抗體診斷試劑盒、艾滋病病毒抗體診斷試劑盒、梅毒抗體診斷試劑盒、丙氨酸氨基轉移酶試劑盒、血型試劑盒、快速篩查試劑盒等。
6.3.9.2 檢驗報告
對於國家進行批批檢的試劑盒,必須有國家批批檢報告。國家不進行批批檢的試劑,以生產廠商出具的出廠檢驗報告爲準。
6.3.9.3 外觀檢查:每批抽檢5盒試劑盒。試劑盒包裝應完整,標識清晰,試劑齊全無滲漏。
6.3.9.4 運輸要求
試劑運送途中的溫度必須符合試劑介紹要求,供應商必須提供試劑運輸冷鏈監控溫度記錄。
6.4 關鍵設備質量檢查
6.4.1 強制檢定設備和校準設備
必須定期對關鍵設備進行檢定或校準,除國家強制檢定設備外,其餘設備血站可以依據國家計量檢定規程,由經培訓具有資質的質控人員自行進行,或委託相關計量機構/生產廠商進行。
6.4.2.1.1 檢查頻率:每年監測1~2次,由血站自行或委託離心機廠商進行。
6.4.2.1.2 離心溫度
檢測方法:於離心機工作間隙,把經計量部門標定的溫差電偶溫度計的探頭放入離心腔內,蓋好離心機蓋。10分鐘後觀察離心機溫度表顯示的溫度與溫差電偶溫度計顯示溫度的差值。
6.4.2.1.3 離心時間
檢測方法:使用秒錶對離心機的時間控制進行檢查。把時間控制表調至規定時間,同時啓動秒錶,觀察離心機時間控制表從開始計時到計時停止秒錶所用的時間。即爲時間控制表按規定時間計時所用的實際時間。
6.4.2.1.4 離心轉速
檢測方法:打開離心機前面板,在連接離心轉頭軸上貼一張反光標籤。把轉速控制調到規定轉速值,然後啓動離心機待轉速穩定後,用轉速儀的光束照明反光標籤,觀察轉速儀顯示屏上的轉速值。或採用其它適宜的方法檢測。
注意事項:爲保證檢查人員的安全,檢測時距轉速儀的測量距離不得小於20cm。
6.4.2.2 儲血設備質量檢查
6.4.2.2.2 溫度
質量標準: 儲血設備的溫度應在規定範圍內,各儲血設備的儲存溫度見下表。
儲血設備溫度標準
檢測方法:使用經計量部門標定的溫差電偶溫度計(精確度爲0.1℃)測定儲血設備箱內的溫度。具體布點方式見YY/T0168-2007《血液冷藏箱》。
質量標準:電源發生故障時,報警系統應立即以聲/光方式發出警報。
檢測方法:切斷儲血設備的電源或開啓報警測試按鈕,模擬電源發生故障,此時報警系統以聲/光方式發出警報。或使用其它適宜的方法檢測。
6.4.2.2.4 溫度失控報警系統
質量標準:當儲血設備的溫度超出質量標準範圍時,報警系統應以聲/光方式發出報警。
檢測方法:將儲血設備的報警範圍分別調至低於和高於儲血設備溫度時,報警系統應以聲/光方式發出報警。
6.4.2.3.2 檢測方法和質量標準:可採用化學指示劑法或生物指示劑法進行滅菌效果監測,具體檢測方法和質量標準見《消毒技術規範》 “壓力蒸氣滅菌效果的監測方法”。
6.4.2.4.2 混勻器搖動頻率
檢查方法:開啓採血秤混勻器後,使用秒錶計時,觀察1分鐘內混勻器搖動次數,搖動一個循環爲一次。
6.4.2.4.3 稱量準確度
質量標準:標示量±2%。
檢查方法:開啓採血秤,將標準砝碼(模擬常規血液採集的重量)置於採血秤上,觀察採血秤顯示的數值。
6.4.2.4.4 報警功能
9 附錄A 獻血者血紅蛋白檢測(硫酸銅目測法)
A.1 試驗原理
全血滴入標準硫酸銅溶液中,形成一層銅蛋白膜,包圍在血滴外層。觀察全血在已知相對密度(比重)硫酸銅溶液中沉浮情況,可判定其相對密度(比重),從而確定其血紅蛋白濃度。
A.2 試劑
A.2.1 用於男性獻血者(Hb≥120g/L)檢測的硫酸銅溶液比重爲1.0520。
A.2.2 用於女性獻血者(Hb≥115g/L)檢測的硫酸銅溶液比重爲1.0510。
A.2.3 不同用途試劑瓶標識應便於區分
A.2.4 採用商品化硫酸銅試劑的,按試劑介紹規定的條件儲存,在有效期內使用。
A.2.5 硫酸銅溶液也可自行配製,配製方法見A.5,應保存配製記錄。
A.2.6 硫酸銅溶液比重隨溫度變化而變化的,溫度每上升2℃,比重下降0.0005。在配製和使用時應注意這一特性。
A.2.7 每瓶(50 ml)硫酸銅溶液的可用於檢測25人次。
A.3 標本要求
A.3.1 新鮮採集的末梢血或靜脈血,抗凝或不抗凝血液均可使用。
A.4 操作步驟和結果判斷
A.4.2 在距硫酸銅液麪上方1cm處垂直將1滴血液輕輕滴入,血滴應不含氣泡。
A.4.3 在15秒內肉眼觀察,判斷結果:1)血滴很快下沉於瓶底,表明血液比重大於硫酸銅溶液比重,血紅蛋白含量符合獻血標準;2)血滴在硫酸銅溶液中部懸浮10 -15秒後下沉於瓶底,表明血液比重等於硫酸銅溶液比重,血紅蛋白含量符合獻血標準;3)血滴懸浮在硫酸銅溶液上部,15秒時不下沉,表明血液比重小於硫酸銅溶液比重,血紅蛋白含量不符合獻血標準。
A.4.4 記錄檢測結果。
A.5.1 貯存液(比重爲1.1000)配製 稱取硫酸銅結晶(CuSO4?5H2O,分析純)170.0g置於大試劑瓶中,在1000ml容量瓶中加入蒸鎦水至刻度,測量此蒸鎦水的溫度,根據表A-1查出與溫度相應的蒸鎦水毫升數,不足的容量用滴定管補加入容量瓶中。將容量瓶中蒸鎦水全部傾入盛硫酸銅結晶的大試劑瓶中,再將容量瓶倒置2分鐘,使蒸鎦水全部滴入大試劑瓶中。此時,容量瓶內尚附着蒸鎦水約0.8ml,在計算時此容量已加在表內數字中。搖動試劑瓶使硫酸銅完全溶解,即得比重爲1.1000的貯存液。如欲知所配製的貯存液比重是否準確,可用液體比重天平進行測定。如配得的貯存液比重並非1.1000,比重每超過0.0001,則在1000ml溶液中加入蒸鎦水1ml。例如,所配成的貯存液的比重1.1050,則在此溶液1000ml中加入蒸鎦水50ml,即可校正其比重爲1.1000。反之,如所配溶液的比重小於1.1000,則每少0.0001,應在1000ml硫酸銅溶液中加入飽和硫酸銅溶液1ml。在盛貯存液的試劑瓶上貼好標籤,做好配製記錄。
表A-1 配製比重1.1000硫酸銅溶液需用蒸鎦水的容量
蒸鎦水溫度℃ | 蒸鎦水量(ml) | 蒸鎦水溫度℃ | 蒸鎦水量(ml) |
10 | 1003.6 | 26 | 1006.5 |
12 | 1003.8 | 28 | 1007.0 |
14 | 1004.0 | 30 | 1007.7 |
16 | 1004.3 | 32 | 1008.3 |
18 | 1004.7 | 34 | 1008.9 |
20 | 1005.1 | 36 | 1009.6 |
22 | 1005.5 | 38 | 1010.4 |
24 | 1006.0 | 40 | 1011.2 |
A.5.2 工作液配製
A.5.2.1 配製100 ml不同比重工作液所需貯存液容量見表A-2。
A.5.2.2 確定工作液配製總量,量取所需量貯存液,加入容器內,慢慢加入蒸鎦水至工作液總量,邊加邊搖勻,以消除硫酸銅溶液加水後容量縮減的影響。配製時貯存液和蒸鎦水的溫度應相同,但不一定爲25℃,10~40℃之間影響不大。
表A-2 硫酸銅應用標準液配製
比重 | 貯存液用量(ml) | 比重 | 貯存液用量(ml) | 比重 | 貯存液用量(ml) |
1.0350 | 34.30 | 1.0490 | 48.20 | 1.0630 | 62.30 |
1.0360 | 35.30 | 1.0500 | 49.20 | 1.0640 | 63.35 |
1.0370 | 36.30 | 1.0510 | 50.20 | 1.0650 | 64.40 |
1.0380 | 37.25 | 1.0520 | 51.25 | 1.0660 | 65.40 |
1.0390 | 38.20 | 1.0530 | 52.25 | 1.0670 | 66.40 |
1.0400 | 39.20 | 1.0540 | 53.30 | 1.0680 | 67.40 |
1.0410 | 40.20 | 1.0550 | 54.30 | 1.0690 | 68.40 |
1.0420 | 41.20 | 1.0560 | 55.30 | 1.0700 | 69.40 |
1.0430 | 42.20 | 1.0570 | 56.30 | 1.0710 | 70.40 |
1.0440 | 43.20 | 1.0580 | 57.30 | 1.0720 | 71.45 |
1.0450 | 44.20 | 1.0590 | 58.30 | 1.0730 | 72.50 |
1.0460 | 45.20 | 1.0600 | 59.30 | 1.0740 | 73.50 |
1.0470 | 46.20 | 1.0610 | 60.30 | 1.0750 | 74.50 |
1.0480 | 47.20 | 1.0620 | 61.30 |
A.5.2.3 工作液配製後應檢測其是否符合質量要求:1)測定比重是否準確,可用液體比重天平測定,必要時採用相對密度測定法;2)採用已知低於和高於獻血者血紅蛋白標準的全血進行檢測,應符合預期要求。
A.5.2.4 將配置完畢的工作液分裝至小試劑瓶,粘貼標籤。
A.5.2.5 記錄配製過程。
10 附錄B 血液檢測方法的確認
B.1 總則
本附錄討論了血站血液檢測實驗室採用一項新的檢測方法,或更換現有檢測方法時需要考慮的因素。通常血液檢測方法包括完成檢測必需的儀器、試劑、校準品、實驗程序組合。檢測方法必須由實驗室選擇。必須按照生產商的介紹進行操作,確保檢測結果符合實驗室的要求。
血站對獻血者及其捐獻的血液進行強制性血液篩查,包括輸血相關感染病原學標誌物檢測、血型血清學檢測和酶學檢測。方法學包括ELISA法、凝集法、速率法等,以及用於獻血者採血前檢查的快速診斷實驗。
由於以上方法學原理的不同,方法確認的原則也不同。依據當今國際公認的一些法規和指南,本附錄提供了血站血液檢測實驗室常規使用的檢測方法確認的原則和步驟,詣在提高血液檢測實驗室對檢測方法確認的一致性和效率,確保檢測方法變化的過程得到有效控制。幫助用戶滿足文件和法規的要求。
B.2 範圍
爲血站血液檢測實驗室檢測方法確認提供方案。實驗室需選擇國家批准的符合國家要求的儀器、試劑盒或檢測方法。在新方法投入常規使用報告檢測結果之前,需要就檢測方法在實驗室的使用性能進行確認。不建議實驗室對生產商提供的檢測方法進行自行改動,如果確實需要修改,應與生產商合作進行新檢測方法性能的評價測試,以確認修改後的系統在實驗室的適用性和可靠性。
需確認的對象包括:
實驗室首次引入的檢測方法,如實驗室從未使用過的試劑、儀器等。
實驗室首次將某項目引入現用檢測方法,如HBsAg項目由A儀器檢測改爲由B儀器檢測。
若多臺儀器(相同品牌和型號)檢測同一個項目,應對每一臺儀器的性能進行確認比較。
以下內容適用於酶聯免疫吸附試驗的確認。快速診斷試驗和確證試驗等定性方法的確認可參照本章節內容。
B.3.1 確認的一般要求
B.3.1.1 實驗操作的培訓
開始確認新的檢測方法之前,操作人員應當有充分的時間熟悉新系統,確定關鍵的操作步驟。生產商應提供試劑或儀器的操作說明,實驗原理,規格,實驗步驟,侷限性,質量控制和健康安全信息。生產商應對操作者進行培訓,確保操作人員正確操作。
B.3.1.2 試劑的準備
應選擇經國家檢定合格的病原學標誌物診斷試劑。嚴格遵從生產商的介紹進行操作。對於正在進行確認的試劑,所有組份應來源於同一批試劑,不能由其他試劑所替代或實驗室自己配製。
B.3.1.3 儀器的準備
應選擇國家批准使用的檢測儀器。實驗開始前,按要求對儀器進行維護校準。對於開放試劑的儀器設備,特別注意試劑介紹要求的操作條件與儀器性能是否相適應。如果儀器性能與試劑盒介紹的要求有差異,可以徵得試劑生產商協助和同意,對本實驗室儀器的實驗操作參數進行修正,以達到試劑盒要求的實驗狀態。
B.3.1.4 質量控制
應建立有效的質量控制程序。除直接採用生產商提供的質控品外,還應選擇質量、來源穩定,無基質效應的質控品作爲另外的室內質控物。如可能,多實驗方法比較過程中,應採用相同質控物。一些定性實驗每天只需要使用陰性和陽性質控物,而另一些定性實驗可以產生量化的質控結果。對此可以採用統計過程控制方法(SPC),用量化數據監控實驗過程。確認實驗採用的標本不能等同於質控品使用。實驗過程中,應確保過程處於受控狀態,否則必須重新實驗。
B.3.1.5 保持記錄
應記錄和監控操作人員在實驗過程中的健康安全,確保符合法律要求。保留檢測全過程的數據和結果,包括檢測的速度和使用的適宜性等。
B.3.2 重複性實驗
B.3.2.1.1 實驗程序:採用生產商提供的陰性、陽性對照,在不低於10天實驗運行的前提下,進行至少20次檢測。
B.3.2.1.2 可接受標準:20次檢測結果不能出現大於1次陰性陽性對照不符合生產商要求。否則實驗室必須終止實驗,諮詢試劑生產商,查找原因,採取糾正措施。
B.3.2.2 方法精密性實驗
B.3.2.2.1 實驗程序:在定性實驗中,如果能夠產生量化實驗結果(如ELISA實驗),應採用接近臨界值的標本,對精密度進行估計。建議採用S/CO爲2~4的標本,不適宜採用低值的陰性標本和高值陽性標本,因爲此類標本距決定水平點即臨界值(cutoff)的分析濃度相距較遠。
B.3.2.2.2 可接受標準:批內變異係數在15%以內,批間變異係數在20%以內。
本實驗採用已知真實血清學狀態的標本,經檢測獲得方法靈敏度和特異性方面的信息。也可以將不同方法的靈敏性和特異性進行比較,得到量化的差異結果,判斷是否具有統計學意義。
B.3.3.1 標本的選擇
靈敏度和特異性實驗通常對已知真實血清學狀態的標本進行檢測。確定標本真實血清學狀態可以採用金標準方法或臨牀診斷方法。參考血清盤或能力驗證(PT)的標本可用於本實驗。參考血清盤是指經過檢測或經過與公認方法比較,或具有臨牀診斷意義的標本。這些標本已經確立了真實的血清學狀態,可追溯至經典方法或臨牀診斷結果。可以從權威機構、專業研究機構和文獻中獲得參考血清盤的來源。參考血清盤應包含具有不同分析物濃度的臨牀標本。如可能,血清盤應包含對檢測具有干擾的標本。如可造成假陰性和假陽性結果的自身免疫性疾病、螺旋體疾病、白細胞抗體、風溼性關節炎、多發性骨髓瘤患者的標本。儘管商品化參考血清盤在方法確認過程中,具有很好的時間和資源效率,但其侷限性在於可能忽視了實驗室目的檢測人羣中疾病的流行率和病原學因子抗原譜的分佈。因此,使用參考血清盤、能力驗證標本結合常規實驗室檢測標本,可以提供更有意義、更可靠的確認結果。
PT材料可能存在基質干擾,對方法性能產生錯誤判斷。當PT材料爲臨界標本,或分子結構不同於天然血清,導致抗原決定簇不能被正常識別時,這種干擾尤其明顯。
B.3.3.2 標本的數量
標本數量的估算,統計學中稱爲標本含量的估算依據實驗設計的類型、結果變量的性質、研究目的和採用的統計分析方法而不同。對於檢測結果爲計數資料的實驗(如定性實驗),一般而言,在實驗誤差控制較好的情況下,不同血清學狀態的標本數量至少爲30份。否則可能影響實驗的統計學功效。
應確保標本可用於常規檢測並處於穩定狀態,儘可能保持實驗標本新鮮。如需凍存,血清或血漿應保存在-20℃的條件下。實驗標本的保存狀態、凍融次數、使用次數和時間應儘量一致,避免標本產生偏差。如需要方法比較實驗,應在相同時間進行,最大限度減小由於標本放置時間不同產生的結果差異。
B.3.3.4 數據的收集和整理
及時記錄和檢查所有實驗數據,儘早發現系統和人爲的誤差。如果能夠確定一些結果是由於可知的差錯造成,應詳細記錄差錯原因,數據不能用於後續分析過程。如果不能夠確定產生異常結果的原因,在數據組中保留原始結果。
B.3.3.5 數據的分析
如果明確了每份檢測標本真實的血清學狀態,檢測方法的靈敏度和特異性很容易計算。表B-1爲反映一個定性實驗檢測已知真實血清學狀態標本的結果的2×2四格表資料。通過這組數據可以計算被估計的靈敏度、特異性。
表B-1 試驗方法結果與診斷結果的2×2四格表
估計靈敏度=100%〔A/(A+C)〕
估計特異性=100%〔D/(B+D)〕
可採用完全隨機設計和配對設計實驗,比較兩檢測方法的靈敏度和特異性。完全隨機設計時,可採用完全隨機設計x2檢驗。兩方法採用不同的標本,標本相對獨立。這種情況下兩方法靈敏度和特異性的比較實際是兩樣本率的比較。配對設計時,可採用配對設計的McNemarx2檢驗。兩方法採用相同的標本,得到兩個相關的靈敏度和特異性,這時注意當標本數較少時,需要計算校正x2或採用精確概率法檢驗。詳細計算見B.3.3.5.3。
注意如果需要判斷檢測方法的準確性,單憑比較靈敏度和特異性是不夠的。除非一個方法的靈敏度和特異性均大於或小於另一方法,否則只能比較相同靈敏度時的特異性,或相同特異性時的靈敏度。當靈敏性和特異性均不相同時,很難比較兩方法準確性。這時可採用受試者工作特徵曲線法(ROC曲線法)進行比較。該法是近年來公認的評價和比較檢測方法準確度的較好方式。該方法可以考慮檢測方法在所有臨界值時的靈敏度和特異性。通過比較不同方法ROC曲線下的面積,確定和參考方法的特性。鑑於此項內容不在本規程要求的範圍,實驗室人員如需要瞭解更多信息,可以查找相關資料。
如果需要量化兩方法靈敏度和特異性之間差異,可以對靈敏度或特異性的差異進行估計。以配對設計實驗爲例,兩方法採用相同的標本進行檢測的結果可以彙集成表B-2。如果兩方法採用的標本不同,則不能採用表2的方式整理數據。
靈敏度1=100%[(a1+b1)/n1]
靈敏度2=100%[(a1+c1)/n1]
特異性1=100%[(c2+d2)/n2]
特異性2=100%[(b2+d2)/n2]
通常採用配對設計四格表x2檢驗和Kappa檢驗評價兩個定性檢測方法性能的差異或一致性。
如果已知標本的血清學狀態,或者已存在特設或隱含的金標準結果,可採用配對設計四格表x2檢驗中的McNemarx2檢驗評價兩方法結果的不一致性,Kappa檢驗評價兩方法結果的一致性。
如果未知標本的血清學狀態,檢測結果有序變量的檔次劃分也是相同的,也可以採用Kappa檢驗評價兩種檢測方法結果的一致性。另外,兩方法的符合性指標也能夠量化兩種檢測方法結果的一致程度。
B.3.3.5.3.1 配對設計四格表x2檢驗(McNemarx2檢驗):對於隱含金標準或特設金標準的2×2四格表資料,可採用McNemarx2檢驗,判斷兩種檢測方法結果不一致部分是否具有統計學意義。該法適用於標本含量適中的資料,因爲本法僅僅考慮兩檢測方法不一致的情況,而未考慮兩方法檢測結果一致的情況。當n很大,兩方法一致率較高,不一致結果數量較小,即使McNemarx2檢驗具有統計學意義,但實際意義往往不大。通常適用於b+c≥40。

注意當25≤b+c<40,採用McNemar檢驗分析配對設計四格表資料需作連續性校正。

B.3.3.5.3.2 Kappa檢驗:Kappa統計量是Cohen1960年提出的一種校正機遇之後衡量兩種檢測方法一致性的指標。Kappa(常縮寫成K)值在-1到+1之間,如果K=-1,說明兩種檢測方法檢測的結果完全不一致;K=0,說明觀察的一致性完全由偶然誤差造成;K=1,說明完全排除機遇一致性後的真正一致性。一般對總體而言,若K>0.75,表明一致性爲優;若K在0.4~0.75之間,表明一致性良好;若K<0.4,表明一致性較差。

如果需要量化兩方法檢測結果的符合程度,可以對兩方法檢測結果的符合性進行估計。

B.3.3.5.3.4 符合性=100%[(a+d)/n]
以下內容適用於ALT速率法確認。其它定量檢測方法確認可參照本規程。
B.4.1 確認的一般要求
B.4.1.1 實驗操作的培訓
開始確認新的檢測方法之前,操作人員應當有充分的時間熟悉新系統,確定關鍵的操作步驟。生產商應提供試劑或儀器的操作說明,實驗原理,規格,實驗步驟,侷限性,質量控制和健康安全信息。生產商應對操作者進行培訓,確保操作人員正確操作被確認系統。
B.4.1.2 試劑的準備
應選擇國家批准生產的ALT診斷試劑。嚴格遵從生產商的介紹進行操作。對於正在進行確認的試劑,所有組份應來源於同一批試劑,不能由其他試劑所替代或實驗室自己配製。
B.4.1.3 儀器的準備
應選擇國家批准使用的檢測儀器。實驗開始前,按照要求對儀器進行校準。校準品必須能夠溯源至國家或國際標準。可以由校準品的生產商提供相關的校準程序和溯源證明。對於開放試劑的儀器設備,特別注意試劑介紹要求的操作條件與儀器性能是否相適應。如果儀器性能與試劑盒介紹的要求有差異,可以徵得試劑生產商協助和同意,對本實驗室儀器的實驗操作參數進行修正,以達到試劑盒要求的實驗狀態。
B.4.1.4 質量控制
應建立有效的質量控制程序。選擇質量、來源穩定,無基質效應的質控品作爲室內質控物。確認實驗採用的標本不能等同於質控品使用。應確保實驗過程處於受控狀態,否則必須重新實驗。
B.4.1.5 保持實驗記錄
應記錄和監控操作人員在實驗過程中的健康安全,確保符合法律要求。保留檢測全過程的數據和結果,包括檢測的速度和使用的適宜性等。
B.4.2 正確度的估計
方法比較實驗:採用新的檢測方法和參加過能力驗證的檢測方法或公認的參考方法,同時檢測一批不同ALT濃度的標本。通過結果的差異瞭解新系統引入後的偏倚是否在允許誤差範圍內。通常將新系統方法稱爲比較方法(y),與之比較的系統方法稱爲參考方法(x),該實驗也稱爲方法學比較實驗。
能力驗證活動:直接參加權威機構組織的能力驗證活動,能力驗證的成績可以證明新的檢測方法檢測結果的準確性。
B.4.2.1 標本的選擇
B.4.2.1.1 標本數量:實驗標本數至少40份,更多的標本量可以使實驗結果更加可靠。每份標本應有足以完成雙份測定的量。如果不夠,可以將分析物濃度接近的兩份標本混合使用。
B.4.2.1.2 標本分析物濃度範圍:如可能,應選擇有50%實驗標本的分析物濃度不在參考範圍內。
B.4.2.2 實驗程序
時間至少5天。採用兩種方法,將標本按照序號遞增和遞減的順序測定2次。如第1次檢測序號爲1、2、3、4、5、6、7、8,第2次爲8、7、6、5、4、3、2、1。當天的方法比較實驗應在2小時內完成。
B.4.2.3 數據分析
B.4.2.3.1 初步判斷:剔除有明顯人爲誤差的結果。內部質控失控時,必須重新檢測。
B.4.2.3.2 離羣點的剔除:設參考方法測定結果爲x值,第一次測定值爲x1,第二次測定值爲x2。比較方法測定結果爲y值,第一次測定值爲y1,第二次測定值爲y2。

以4 和4
和4 爲離羣限值,如果沒有超出,繼續方法間配對結果離羣點檢驗;否則繼續相對差異檢驗,進行如下最終判斷
爲離羣限值,如果沒有超出,繼續方法間配對結果離羣點檢驗;否則繼續相對差異檢驗,進行如下最終判斷

最終以4和4爲判斷離羣點限值
B.4.2.3.4 方法間配對結果離羣點的去除
計算Eij=∣yij-xij∣,i爲標本序號1~40,j爲重複結果序號1或2。
計算
以4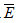 爲離羣限值。如果全部實驗數據中離羣點超過2個,應補充實驗數據。
爲離羣限值。如果全部實驗數據中離羣點超過2個,應補充實驗數據。
x-y散點圖
以參考方法所有結果爲x值,比較方法所有結果爲y值,繪製散點圖。
以參考方法每個標本兩次檢測結果的均值爲x值,比較方法每個標本兩次檢測結果的均值爲y值,繪製散點圖。比較兩圖有無明顯差異。如無明顯差異,說明方法比較實驗的隨機誤差對比較結果影響不大。
x均值(x-y)均值差散點圖
以參考方法每個標本兩次檢測的均值爲x值,兩方法實驗結果均值差(x-y)爲y值,繪製散點圖。觀察均值差隨x值增大的變化。如果點的分佈表現出一定的趨勢,預示着兩方法之間系統誤差的存在。
實驗點的離散度和對應分析物濃度分佈的寬度都會影響r值的大小。一般如果r≥0.975(r2≥0.95),x取值範圍適宜,否則應擴大數據範圍。
B.4.2.3.7 線性迴歸計算
每份標本用兩個方法進行兩次測定。將參考方法的x1和比較方法的y1對應,參考方法的x2和比較方法的y2對應,組成成對數據。採用所有成對數據進行直線迴歸。得迴歸方程 。理想狀態下 ,即b=1, =0。如果b≠1, ≠0,說明兩方法間存在系統誤差,需要對誤差進行評估。如瞭解Y方法引入後相對於X方法在臨牀決定水平濃度Xc處的系統誤差(SE)。
。理想狀態下 ,即b=1, =0。如果b≠1, ≠0,說明兩方法間存在系統誤差,需要對誤差進行評估。如瞭解Y方法引入後相對於X方法在臨牀決定水平濃度Xc處的系統誤差(SE)。
B.4.2.4 可接受標準
r≥0.975或r2≥0.95。醫學決定水平處新方法引入的系統誤差小於CLIA’88允許誤差的1/2。
B.4.3 精密度的估計
精密度估計最常用的方法是採用穩定的標本進行重複檢測。求得均值(x)、標準差(s)和變異係數(CV)。精密度的好壞通常採用不精密度表示,不精密度通常以CV體現。應在明確標本的來源、濃度、實驗週期的前提下,確定檢測方法的不精密度。
B.4.3.1 標本的選擇
B.4.3.1.1 標本分析物濃度範圍:鑑於血站血液檢測實驗室檢測對象爲健康的獻血人羣,可採用中、低兩個ALT濃度的標本進行精密度實驗。標本的基質應近似於人源標本。
B.4.3.1.2 標本的保存:精密度實驗需要在若干天內完成,對於冰凍保存的標本和凍幹復溶標本,應嚴格控制操作手法均一、復溶時間、加液的準確性、混勻的力度等,力求實驗標本的穩定、均一。
B.4.3.2 實驗程序
B.4.3.2.1 對每個水平的標本分別做批內和天間的重複測定。精密度實驗與常規標本檢測同時進行,不可替代實驗的質量控制過程。
B.4.3.2.2 批內實驗:將實驗標本插入常規檢測標本一同檢測。一批內連做20次。對應質控結果爲受控狀態。
B.4.3.2.3 批間實驗:每天對實驗標本做1次檢測,連做20天。每天對應質控結果爲受控狀態。
B.4.3.3 數據分析
B.4.3.3.1 標準差計算
批內和天間標準差均可採用以下公式計算

B.4.3.3.2 變異係數計算
批內和天間變異係數均可採用以下公式計算

B.4.3.4 可接受標準
參照Westgard和Ceroblewski等提出高效檢驗對精密度要求的觀點,批內不精密度CV應小於CLIA允許誤差的1/4,批間不精密度CV應小於CLIA允許誤差的1/3。對於ALT實驗,CLIA允許誤差限值T±20%,因此ALT檢測方法的批內不精密度CV應小於5%,批間不精密度CV應小於6.7%。
B.4.4 檢測可報告範圍
檢測可報告範圍確認實驗通常採用配製包含不同分析物濃度的系列標本進行檢測,將按稀釋比例計算的預期值和實際測量值設置爲兩個變量進行線性迴歸。迴歸直線應當爲過原點,斜率爲1的直線。直線所達的限值即爲結果的檢測範圍。
B.4.4.1 標本的選擇
準備一份ALT<30IU的低濃度血清(L)和ALT在500IU左右的高濃度血清(H)。將高低兩份血清按照5L, 4L+1H, 3L+2H, 2L+3H, 1L+4H, 5H的比例配製混合,形成系列血清。
B.4.4.2 實驗程序
B.4.4.3 數據分析
B.4.4.3.1 剔除異常點:剔除明顯異常的檢測結果,並補充數據。
B.4.4.3.2 計算預期值:計算低值和高值標本的結果均值。按照稀釋關係,計算每個標本的分析物理論濃度,即爲預期值。
B.4.4.3.3 預期值-實測值的線性迴歸:每個標本有4個重複實驗的實測值。每個實測值與其預期值相對應,形成成對數據。以x表示預期值,y表示實測值,描點繪圖。得到迴歸方程 。
。
B.4.4.3.4 初步判斷:理想狀態下中b=1,a=0。但實際工作中很難達到。因此如果b在1.00±0.03的範圍內,a近於0,則可直接判斷該方法可報告範圍在實驗涉及濃度範圍內。
B.4.4.3.5 如果b不接近1,a較大,應具體分析高濃度還是低濃度的預期值和實測值存在較大偏倚。可以捨去某組數據,重新進行迴歸計算。如果縮小範圍後,b和a能夠滿足要求,即說明縮小的濃度範圍即是實際的檢測範圍。
B.4.4.3.6.1 截距a和0差異的t檢驗
假設H0:a=0;H1:a≠0;α=0.05

B.4.4.3.6.2 斜率b和1差異的t檢驗
假設H0:b=1;H1:b≠1;α=0.05

B.4.5 正常值參考範圍確認
採用新方法對20份健康獻血者血液標本進行檢測。最多允許1個結果不在試劑盒提供的參考範圍內。
B.5.1 確認的一般要求
B.5.1.1 實驗操作的培訓
開始確認新的檢測方法之前,操作人員應當有充分的時間熟悉新系統,確定關鍵的操作步驟。生產商應提供試劑或儀器的操作說明,實驗原理,規格,實驗步驟,侷限性,質量控制和健康安全信息。生產商應對操作者進行培訓,確保操作人員正確操作。
B.5.1.2 試劑的準備
應選擇國家批准使用的血型試劑,嚴格遵從生產商的介紹進行操作。對於正在進行確認的試劑,所有組份應來源於同一批試劑,不能由其他試劑所替代(即使另一種試劑爲同一廠家生產)或實驗室自己配製。實驗室對試劑自行修改,如稀釋試劑,應取得生產商的認可,因爲任何修改可以導致對試劑的質量保證失效,併產生相關責任。
B.5.1.3 儀器的準備
應選擇國家批准使用的檢測儀器。實驗開始前,按照要求對儀器進行維護校準。對於開放試劑的儀器設備,特別注意試劑介紹要求的操作條件與儀器性能是否相適應。如果儀器性能與試劑盒介紹的要求有差異,可以徵得試劑生產商協助和同意,對本實驗室儀器的實驗操作參數進行修正,以達到試劑盒要求的實驗狀態。
B.5.1.4 質量控制
應建立有效的質量控制程序。確保常規平行實驗中已知抗原和已知抗體的質控物反應正確,否則必須重新實驗。
B.5.1.5 保持實驗記錄
應記錄和監控操作人員在實驗過程中的健康安全,確保符合法律要求。保留檢測全過程的數據和結果,包括檢測的速度和使用的適宜性等。
B.5.2 平行確認實驗
所有實驗室應採用平行實驗的方式,用常規標本,將新方法與現用方法進行比較,確認新方法的常規使用狀態。實驗時間最短1周。操作程序和實驗結果應文件化。應定期進行關鍵標本(弱抗原或弱抗體標本)重複實驗,確認方法的穩定性和耐用性。
B.6 確認記錄
B.6.1 實驗過程記錄應包括:
B.6.1.2 檢測日期;
B.6.1.3 操作實驗者的級別和能力;
B.6.1.4 使用試劑的詳細資料,包括:批號、效期等;
B.6.1.5 採用修正條件的詳細資料,如溫度、孵育時間,離心力、加速減速率、離心時間;
B.6.1.6 設備詳細資料;
B.6.1.7 判讀方法的詳細資料,包括適當的cutoff值。
B.6.2 實驗結果記錄應包括:
B.6.2.1 所有結果並繪製表格,包括陽性反應和陰性控制結果;
B.6.2.2 差異結果、數量和類型,以表格形式體現。
B.6.3 確認結論記錄應包括:
B.6.3.1 用戶界面友好性;
B.6.3.2 操作人員需要的水平,培訓要求;
B.6.3.4 不同情況下使用的適宜性(批量標本處理或單一緊急標本處理);
B.6.3.6 方便的儀器維護;
B.6.3.7 健康安全方面,包括數據採集和處理安全性;
B.6.3.8 任何過程中暴露的系統弱點和對系統改進的建議,並通知生產商,如果生產商對此實施了改進方案,重新實驗的結果應在報告中體現。
11 附錄C 血液檢測試劑(酶聯免疫/核酸試劑)
進貨驗收與放行記錄表
1 訂購 試劑品名 規格 生產方 |
銷售方 銷售員姓名及身份證號碼 |
試劑產銷證照是否齊全有效? 採購合同是否有效? |
訂購數量 訂購價格 訂購者 訂購日期 |
2 驗貨與隔離 驗貨結論 倉管員 驗貨日期 |
2.1 外觀檢查(箱體完整無損,運輸冷鏈符合要求,試劑包裝盒完整無損,無液體泄漏) |
4 測試 測試結論 測試者 日期 |
4.1 試劑介紹版本檢查 |
4.2 試劑盒組成、組分性狀與介紹一致;無泄漏,足量,標識正確 |
4.3.3 其它檢測要求: |
5 審批 |
(1)准予放行使用。 實驗室主管: 日期: 質量部門主管: 日期: |
(2)退貨。 實驗室主管: 日期: 質量部門主管: 日期: |
6 放行 ①“可用”標識;② 賬物入合格品庫;③ 向BMIS錄入試劑信息 倉管員 日期: |
7 附件:① 銷售員身份證複印件;② 出廠質量檢驗報告;③ SFDA批簽發文件;④ 試劑介紹;⑤ 測試記錄 |
8 附註 :國家食品藥品監督管理局簡稱SFDA |
12 附錄D 血液檢測室內質控方法
D.1 總則
血站血液檢測主要包括用於血清學抗體或抗原檢測的ELISA試驗,用於HBV/HCV/HIV DNA或RNA檢測的NAT試驗,以及ALT酶學試驗。ELISA試驗室內質控通常採用試劑盒陰陽性對照、弱陽性質控品實時監控實驗的有效性,同時採用弱陽性質控品和Levey-Jennings質控圖監控實驗的穩定性。ALT作爲定量試驗通常採用Levey-Jennings質控圖監控ALT實驗的精密性和有效性。NAT試驗以及其它定性試驗可借鑑ELISA 的質控方法。
推薦採用Levey-Jennings質控圖監控ELISA實驗過程的穩定性,發現隨機誤差和系統誤差。實驗室如需採用更多種類的控制圖進行質控,可查閱相關文獻。
D.2.1 質控規則
D.2.1.1 質控規則的表示方法
用AL方式表示質控規則,“A”代表質控測定值個數,“L”是從正態統計量得到的質控界限。例如,13S質控規則指一個質控結果超出了均值加減3倍標準差界限。
D.2.1.2 質控規則的選用
實驗室可選擇Levey-Jennings質控圖常規使用的13S規則作爲在控與失控的判斷規則,如發現違背13s規則的情況,說明實驗過程沒有處於受控狀態,應查找原因予以解決。實驗室應根據實際情況,同時選擇一個監控實驗系統誤差的規則,如7x規則,以發現由於儀器、試劑、環境條件等因素引起的系統誤差。上述常用質控規則的釋義如下:
13s:一個質控值超過 。用於提示可能存在隨機誤差。
。用於提示可能存在隨機誤差。
7x:7個連續的質控值落在均值一側,用於提示可能存在系統誤差。
D.2.2 質控圖的建立
在實驗室常規檢測條件下,連續測定同一批號的弱陽性質控品10~20天,收集至少20個質控數據,對數據進行離羣值檢驗,剔除超過±3s以外的數據,計算均值及標準差,以此控制後續實驗過程,直至試劑或質控品批號更換。實驗室如採用積累質控數據計算均值和標準差,具體方法可參見本章節ALT質控均值的計算。但需注意,由於ELISA試劑存在較大的批間差異,積累質控數據計算的方式可能增大室內質控的標準差和變異度,因此如兩批試劑的質控均值和標準差有顯著差異,建議針對新批號試劑重新計算質控均值和標準差。
實驗室構建質控圖的過程中,如果發現實驗變異度過大,應採取措施,穩定各個環節實驗條件,將變異度控制在可接受範圍內。通常情況下,ELISA實驗的變異係數宜控制在20%以內。
控制限通常是以標準差的倍數來表示。Levey-Jennings質控圖將 設置爲控制限,即控制上限值爲 ,控制下限值爲
,控制下限值爲 。如果質控圖的控制下限值小於1,說明實驗室採用的控制低限(LCL)已經低於實驗性能低限(LSL),實驗過程變異較大,實驗室應當查找原因,改進過程,降低實驗變異度(CV)。
。如果質控圖的控制下限值小於1,說明實驗室採用的控制低限(LCL)已經低於實驗性能低限(LSL),實驗過程變異較大,實驗室應當查找原因,改進過程,降低實驗變異度(CV)。
D.2.2.3 繪製質控圖
以Y軸爲質控品的測定值(S/CO值),X軸爲質控個數或單位時間。Y軸刻度上各水平線分別爲均值 上下限,描點繪圖。
上下限,描點繪圖。
當採用13s規則可採用單點質控圖,將質控數據逐一點於質控圖上進行觀察,以發現實驗隨機誤差。
當採用7x規則,可根據實驗室情況,採用單點質控圖或將一個單位時間內(通常爲一天)所有質控數據的均值點於質控圖上進行觀察,以發現檢測系統的變化和趨勢。
D.2.2.4 質控圖框架的重建
如果更換新批號試劑,鑑於ELISA實驗的特性和試劑批間的不穩定性,可能兩批試劑質控均值和標準差存在顯著差異,如需要應重新建立質控圖框架。
如果更換新批號質控品,在試劑批號不變的情況下,可採用將新批號質控品和舊批號質控品同時檢測的方式,以確保在舊批號質控品使用結束前,獲得計算新批號質控品均值和標準差的數據,建立質控圖框架。
如果質控圖使用過程中,出現均值的偏移和標準差變化,需分析並消除產生偏差的原因,必要時應重新調整質控圖框架。
D.2.3 失控情況的分析處理
如出現違背實驗有效性判定規則,應視爲實驗無效。查找原因,採取糾正措施後重新實驗。
如違背實驗室選擇的質控規則,出現隨機誤差或系統誤差,實驗室應分析產生誤差原因。引起誤差的因素通常包括操作上的失誤,試劑、校準物、質控品的失效;試劑、質控品更換批號或保存末期發生變化;儀器使用維護不當;質控圖建立過程中採用的數據不足造成均值和標準差不適宜等。應採取糾正措施,消除產生誤差的因素。如果所選擇弱陽性質控品超過規定的S/CO值上限,應關注違背-3s規則時的實驗狀況,必要時對陰性結果重新檢測。應保存失控情況分析處理記錄。
D.3 ALT實驗室內質控
D.3.1 質控規則的選用
D.3.2 質控圖的建立
D.3.2.1 設定質控圖均值:在實驗室常規檢測條件下,測定同一批號的質控品10~20天,收集至少20個質控數據,對數據進行離羣值檢驗,剔除超過±3s以外的數據,計算臨時均值及標準差,以此控制此後一個月的室內質控情況。一個月結束後,彙集當月所有質控數據,計算所有在控數據積累的質控均值和標準差,以此控制下一個月的室內質控情況。重複以上操作三到五個月,最終彙集前20個數據和三到五個月的質控數據,計算積累的質控均值和標準差,以此作爲常規均值和標準差,控制以後的實驗過程,直至質控品批號更換。
D.3.2.2 設定質控圖控制限:控制限通常以標準差的倍數表示。Levey-Jennings質控圖將 設置爲控制限,即控制上限值爲
設置爲控制限,即控制上限值爲 ,控制下限值爲
,控制下限值爲 。ALT室內質控的控制限隨質控圖均值和標準差的變化而變化。
。ALT室內質控的控制限隨質控圖均值和標準差的變化而變化。
D.3.2.3 繪製質控圖
D.3.2.4 質控圖的重建:鑑於ALT實驗的特性,質控圖可包含多個試劑批號的變化。如果更換新批號質控品,可採用將新批號質控品和舊批號質控品同時檢測的方式,以確保在舊批號質控品使用結束前,獲得計算新批號質控品均值和標準差的數據,建立質控圖。
D.3.2.5 如果質控圖使用過程中,出現均值的偏移和標準差變化,需分析並消除產生偏差的原因,必要時應重新構建質控圖。
D.3.3 失控情況的分析處理:如出現違背質控規則的情況,需分析產生誤差的類型及原因,並視爲實驗失控和無效。應查找原因,採取糾正措施後重新實驗。引起誤差的因素通常包括操作上的失誤、試劑、校準物、質控品的失效;試劑、質控品更換批號或保存末期發生變化;儀器使用維護不當;由於質控圖建立過程中採用的數據不足造成均值和標準差不適宜等。應保存失控情況分析處理記錄。
D.4 特殊情況下的室內質控
對於不能每天進行血液檢測、質控數據量少的實驗室,必須保證每次實驗滿足試劑盒質控要求,弱陽性質控品S/CO≥1。在次基礎上,可適時採用Grubbs氏法進行室內質控。
該方法只需連續測定3次,即可對第3次檢驗結果進行檢驗和控制。具體計算方法如下。
1)計算出測定結果(至少3次)的平均值( )和標準差(s)。
)和標準差(s)。
2)計算SI上限值和SI下限值:
SI上限 = (X最大值-)/s
SI下限 = (-X最小值)/s
13 附錄E 微板法ABO血型定型試驗
E.1 本附錄描述了基於手工操作或半自動操作的U型微板技術的基本步驟。
E.2.1 在微板的3個乾淨孔內分別加入1滴抗-A、抗-B,(如需要,可在第三孔內加1滴抗-AB進行平行試驗)。
E.2.2 在含有血型定型試劑的各孔內加入1滴1.6%~1.8%紅細胞懸液;輕叩板邊,混勻孔內物質。可以採用自動化的樣本處理系統完成抗血清和受檢紅細胞懸液的配製和加樣。
E.2.3 離心和重懸。離心結束後用手輕叩微板或藉助微板振盪器重懸細胞扣。
E.2.4 肉眼觀察紅細胞是否凝集,判讀結果並記錄,並與反定型結果比較。也可以採用具有血型判讀功能的酶標儀進行結果的自動判讀。
E.3.1 在微板的兩個乾淨孔內,每孔內加1滴受檢者血清或血漿;
E.3.2 在含有血清或血漿的各孔內分別加1滴1.6%~1.8%A1、B細胞(如需要,可添加A2或O細胞進行附加試驗);輕叩板邊,混勻孔內物質。可以採用自動化的樣本處理系統完成受檢血清/血漿和試劑紅細胞懸液的配製和加樣。
E.3.3 離心和懸浮。採用既定的離心條件對微板進行離心。結束後用手輕叩微板或藉助微板振盪器重懸細胞扣
E.3.4 肉眼觀察紅細胞是否凝集,判讀結果並記錄,並與正定型結果比較。也可以採用具有血型判讀功能的酶標儀進行結果的自動判讀。
E.4 結果判讀與解釋
E.4.1 紅細胞溶血或在微板底部形成凝集細胞扣都表示陽性結果;
E.4.3 若正反定型結果不符,應分析不一致的原因,並進一步試驗以確定血型。
E.5 注意事項
E.5.1 如果使用稀釋的抗血清工作液,應關注抗血清試劑的強度和粘度;蛋白濃度過高或過低都會影響檢測結果。稀釋後工作液的保存不應超過1周。
E.5.3 微板使用前可以用0.1%的牛白蛋白或人血清進行預處理以減少塑料的非特異性吸附。加熱洗滌或用溼巾擦拭U型孔底可減少靜電作用。使用後的微板必須徹底清洗方可重複使用。
E.5.4 離心條件和離心後對微板的懸浮條件應標準化。通常軟質U型板離心條件700g×5s;硬質U型板離心條件400g×30s;硬質V型板離心條件700g×10s。懸浮條件應關注懸浮的頻率、幅度和時間。
E.5.5 如果血型試劑未註明可用於微板試驗,或檢測設備爲非系統型,實驗室應對微板法血型定型的各個試驗條件進行優化和驗證。以確保血型檢測的準確性。
14 附錄F 血液質量控制檢查方法
F.1 外觀
F.2 標籤
F.3 容量
F.3.1.1 稱量血袋重量
F.3.1.2 計算公式

注:如果適用,還應減去抗凝劑容量。
F.4 無菌試驗
無菌試驗方法遵從中華人民共和國藥典(2010年版)三部“無菌檢查法”要求。使用細菌培養儀進行無菌試驗應參照廠家使用說明書。
F.4.2 注意事項
留取血液樣本前,應當將血袋中的血液充分混勻,混勻時動作應輕柔,不能產生泡沫,也不能使其溶血。
F.5 Hb測定
檢測方法見《全國臨牀檢驗操作規程》(第3版)“血紅蛋白測定”。使用血細胞計數儀測定的應參照廠家使用說明。
F.6 遊離Hb測定
檢測方法參照《全國臨牀檢驗操作規程》(第3版)“血漿遊離紅蛋白測定”。
F.7 血細胞比容
檢測方法見《全國臨牀檢驗操作規程》(第3版)“紅細胞比容”。使用血細胞計數儀測定的應參照廠家使用說明。
F.8 儲存期末溶血率
在血液儲存期的最後一週內取樣,按 F.5、F.6和F.7測定總血紅蛋白、上清遊離血紅蛋白、血細胞比容。
F.8.2 計算公式

F.9 白細胞殘留量
材料:大容量 Nageotte血細胞計數盤、顯微鏡、結晶紫染色液。
結晶紫染色液配製
儲存液配製:250mg結晶紫溶於250mL的50%醋酸溶液中。室溫放置可保存6個月。
使用液配製:1份儲存液加9份3%的醋酸溶液,最終濃度爲:結晶紫0.01mg%(W/V),醋酸7.7%(V/V)。然後使用0.22μ過濾器過濾。
計數方法:50μl血液標本加入450μl使用液中,充分混勻。在Nageotte計數池中加入上述混合液,將計數池置於帶蓋潮溼容器中,於室溫放置10~15分鐘。在200倍顯微鏡下對計數池中兩個計數區(每區有20個長方形格)的白細胞計數,40行相當於50μl。
計算:

公式中:10爲稀釋倍數,50μl爲計數池2個區的容積
每袋中殘餘白細胞數按下列公式計算:
殘餘白細胞數/袋=白細胞數/mL×去白細胞血液容量(mL)/袋
F.10 紅細胞計數
檢測方法見《全國臨牀檢驗操作規程》(第3版)“紅細胞計數”。使用血細胞計數儀測定的應參照廠家使用說明。
F.11 血小板計數
檢測方法見《全國臨牀檢驗操作規程》(第3版)“血小板計數”。使用血細胞計數儀測定的應參照廠家使用說明。
F.12 血漿蛋白測定
檢測方法見《全國臨牀檢驗操作規程》(第3版)“血清總蛋白的測定”。
F.13 上清液蛋白含量
檢測方法參照《全國臨牀檢驗操作規程》(第3版)“腦脊液總蛋白測定”。
F.14 pH測定
F.14.1 測定方法
測定方法見《中華人民共和國藥典》(2010年版)三部附錄“pH值測定法”。
F.14.2 注意事項
使用國家計量部門檢定合格的pH儀。樣品從血袋中移出後應立即進行檢測,以防標本在空氣中暴露時間過長而CO2逸出,導致pH測定值比實際值偏高。
F.15 Ⅷ因子活性測定
測定方法見《全國臨牀檢驗操作規程》(第3版)“凝血因子Ⅷ的活性測定(一期法)”。使用血凝儀測定時參照廠家使用說明書。
F.16 纖維蛋白原測定
測定方法見《全國臨牀檢驗操作規程》(第3版)“血漿纖維蛋白原含量測定”。使用血凝儀測定應參照廠家使用說明書。根據冷沉澱的容量計算出每袋纖維蛋白原含量。
F.17 甘油殘留量
F.17.1 過碘酸鈉法
原理:根據過碘酸鈉能氧化有機化合物中的羥基、胺基,使甘油氧化成酸和醛,以溴甲酚紫作指示劑;並用氫氧化鈉進行滴定。從消耗氫氧化鈉的毫升數,即可算出樣品中所含甘油的量。
試劑:16.5%鎢酸鈉溶液:稱取16.5克鎢酸鈉,用蒸餾水溶解,再加蒸餾水稀釋至100mL。
0.5mol/L H2SO4:吸30mL濃硫酸緩緩注入水中,冷卻至室溫,加水稀釋至1000mL。
0.1mol/LNaOH:配製方法見《中華人民共和國藥典》(2010年版)二部 附錄 “滴定液”
0.1%溴甲酚紫溶液:0.1g溴甲酚紫溶入20mL0.02mol/L NaOH溶液中,再加入蒸餾水稀釋至100mL。
14%過碘酸鈉:14g過碘酸鈉先加入少量蒸餾水中溶解,加入2mL濃硫酸使其完全溶解,用蒸餾水稀釋至100mL。
操作方法:按下表操作步驟製備血濾液
血濾液製備
注:實驗操作時應在三角燒瓶中進行
分析測定的步驟如下表:
計算:

0.1mol/L NaOH溶液相當於0.00921g甘油
F.17.2 滲透壓測定法
正常血清的滲克分子濃度爲289mmol(289mOsm),甘油含量爲1%(10g/L)時,其滲克分子濃度爲420mmol(420mOsm),因此滲透壓測定法可作爲一種檢測去甘油紅細胞甘油殘存量的參考方法,操作方法應參照滲透壓儀器生產廠家使用說明書。
F.17.3 折射儀測定法
使用折射儀是測定滲克分子濃度的一種篩選方法。折射儀讀數到28(如折射儀型號爲10401,折射指數或比重<1.3384T.S.)與甘油水平不高於1%(90g/L)相關。折射儀測定法可作爲一種檢測甘油含量的參考方法。方法見生產廠家使用說明書。
F.17.4 由於不同方法的準確性和精密度存在差異,可根據要求選擇以上相應方法或其它方法(如:GPO-PAP二步法)。若採用商品化試劑盒,操作應按試劑盒介紹進行。
F.18 中性粒細胞計數
檢測方法見《全國臨牀檢驗操作規程》(第3版)“白細胞計數”。使用血細胞計數儀測定的應參照廠家使用說明。
F.19 亞甲藍殘留量
F.19.1 測試樣品準備
取3mL Waters Oasis小柱,置於真空萃取裝置上,用6mL甲醇活化,加入6mL供試血漿,萃取亞甲藍;用30%甲醇6mL清洗,隨後用含1%乙酸的甲醇溶液2mL洗脫,洗脫液離心(3500rpm/10min),取上清液作爲樣品溶液。
F.19.2 標準品的製備
取亞甲藍粉末約38mg,精密稱定,置100mL量瓶中,用水溶解並稀釋至刻度。精密量取50μl置50mL容量瓶中,用血漿稀釋至刻度,配成最終濃度爲約1.0μmol/L的亞甲藍對照血漿。檢測過濾前後樣品溶液時,必須用同樣的方法萃取檢測標準品。
F.19.3 測定
用含1%乙酸的甲醇溶液作試劑空白,在654nm±2nm波長處測定吸光度。按以下公式計算亞甲藍在血漿中的殘留量:

F.19.4 注意事項
病毒滅活冰凍血漿在留取標本前應置30~37℃融化,融化時間應小於6分鐘。爲使其儘快融化,應不斷輕柔搖動血漿袋。不得急劇搖動血漿袋,以防止血漿中出現大量泡沫。
15 附錄G 血袋標籤確認方法
G.1 範圍
G.2 標籤紙質和粘貼牢固度
G.2.1 質量標準
1)標籤的底色應爲白色,上面的字體建議採用實體黑色字體。
2)全血標籤經離心後,血漿標籤經離心、冷凍、水浴後,標籤不能分離。
3)標籤外觀沒有破損,相對平整。如出現皺褶,不能影響條碼的掃描和讀碼。
4)粘貼在導管上的條碼,經離心、冷凍後在常溫條件下自然融化2小時,條碼與導管之間不能出現分離。
5)對直接粘貼在冰凍血漿(製品表面不平整)袋上的標籤,粘貼時製品不需擦拭,可直接粘貼,不能出現標籤打滑的現象。
對冷藏後血液製品上的標籤,應進行血袋與血袋之間、手指與標籤之間進行摩擦,摩擦後標籤上的油墨不得脫落。
按採供血過程血袋標籤粘貼的操作程序,將抽檢標籤粘貼於規定的部位(血袋上、標籤上或留樣皮管)上,在分離紅細胞和血漿的離心條件下離心30分鐘。離心時將粘貼有標籤的血袋按離心杯的容量整齊地擺放於離心杯中。
將血液和製品需冷凍的抽檢標籤粘貼於規定的部位(血袋上、標籤上或留樣導管)上,放置-18℃低溫冰箱冷凍24小時。
對需經水浴的血液製品標籤應進行水浴試驗。取出粘貼有標籤的冷凍血袋,放入於37℃溫水池中水浴60分鐘(低溫沉澱物製品在2~6℃水浴4小時,再放入37℃溫水池中水浴60分鐘)後取出。
對直接粘貼在冰凍血漿袋上的標籤,應將標籤直接粘貼在冰凍好的血袋上。
對需冷藏的血液製品標籤放入2-6℃的冷藏冰箱,24小時後取出。
G.2.7 注意事項
標籤粘貼牢固度試驗在操作時應將標籤平整地粘貼在血袋上。採供血過程中未規定的標籤粘貼過程不應作爲標籤粘貼牢固度試驗的過程。
G.3 條碼掃描和讀碼
G.3.1 質量標準
各類條碼在常溫、冷藏、冷凍、水浴後均能爲條形碼識讀器所讀碼。
G.3.2 測定方法
各類條碼在常溫、冷藏、冷凍、水浴後用條形碼識讀器讀取。
G.4 印章和書寫字跡清晰度
G.4.1 質量標準
標籤在加蓋印章和油性筆書寫後,印章油印及書寫字跡待幹後不發生嚴重模糊,或部分發生模糊但仍能夠清楚地辯讀。
G.4.2 測定方法
將標籤加蓋印章和使用指定的油性筆書寫,觀察其清晰程度。
16 附錄H(資料性附錄)血站使用的強制檢定工作計量器目錄
H.1 本目錄內項目,凡用於貿易結算、安全防護、醫療衛生、環境監測的,均實行強制檢定。
H.2 本目錄所列的計量器具是根據血站實際工作情況,從《中華人民共和國強制檢定的工作計量器具目錄》節錄而成,並對序號進行了重新編排。包括:
3)磚碼:砝碼;
4)天平:天平;
5)秤:電子秤;
7)密度計:密度計;
10)電離輻射防護儀:射線監測儀、照射量率儀、放射性表面污染儀、個人劑量計;
14)分光光度計:可見光分光光度計、紫外分光光度計、紅外分光光度計、熒光分光光度計、原子吸收分光光度計;
15)比色計:濾光光電比色計、熒光光電比色計;
16)血球計數器:電子血球計數器。