3 概述
核磁共振波譜法是指用波長很長的電磁波,相當於射頻區大於照射分子時,它能夠與暴露在強磁場中的一定原子核相互作用,並且在某些特定磁場強度產生強弱不同的吸收信號,以這種原理建立起來的方法稱爲核磁共振波譜法,或稱核磁共振光譜法,簡稱NMR。以磁場強度或頻率作爲橫座標,以共振信號強度爲縱座標得到核磁共振光譜圖。光譜圖中,共振信號強度直接反映了測定核的數目。另外,由於質子或其他種類核在分子中所處的化學環境不同,在射頻區照射下,產生不同的吸收信號。主要用於結構的測定和確證。
4 核磁共振(NMR)波譜的原理
核磁共振(NMR)波譜是一種基於特定原子核在外磁場中吸收了與其裂分能級間能量差相對應的射頻場能量而產生共振現象的分析方法。核磁共振波譜通過化學位移值、譜峯多重性、偶合常數值、譜峯相對強度和在各種二維譜及多維譜中呈現的相關峯,提供分子中原子的連接方式、空間的相對取向等定性的結構信息。核磁共振定量分析以結構分析爲基礎,在進行定量分析之前,首先對化合物的分子結構進行鑑定,再利用分子特定基團的質子數與相應譜峯的峯面積之間的關係進行定量測定。[1]
帶正電荷的原子核在作自旋運動時,可產生磁場和角動量,其磁性用核磁矩μ表示,角動量P的大小與自旋量子數I有關(核的質量數爲奇數,I爲半整數;核的質量數爲偶數,I爲整數或0),其空間取向是量子化的;μ也是一個矢量,方向與P的方向重合,空間取向也是量子化的,取決於磁量子數m的取值(m=I,I-1,……-I,共有2I+1個數值)。對於1H、13C等I=1/2的核,只有兩種取向,對應於兩個不同的能量狀態,粒子通過吸收或發射相應的能量在兩個能級間躍遷。
當自旋量子數I≠0的磁核處於一個均勻的外磁場H0中時,磁核因受到磁場的作用力而圍繞着外磁場方向作旋轉運動,同時仍然保持本身的自旋。這種運動方式稱爲拉摩進動。原子核的進動頻率由下式決定:
ω0=γH0
其中γ爲旋磁比,是原子核的基本屬性之一。不同原子核的γ值不同,其值越大,核的磁性越強,在核磁共振中越容易被檢測。如果提供一個射頻場,其頻率(ν)滿足:
△E=hν=μH0/I
其中h爲普朗克常數,則:
v=ω0/2π=γH0/2π
即射頻場的頻率正好等於在磁場H0中的核進動頻率,那麼核就能吸收這一射頻場的能量,導致在兩個能級間躍遷,產生核磁共振現象。
核磁共振波譜是一專屬性較好但靈敏度較低的分析技術。低靈敏度的主要原因是基態和激發態的能量差非常小,通常每十萬個粒子中兩個能級間只差幾個粒子(當外磁場強度約爲2T時)。
5 核磁共振波譜儀
常見的有兩類核磁共振波譜儀:經典的連續波(CW)波譜儀和現代的脈衝傅里葉變換(PFT)波譜儀,目前使用的絕大多數爲後者。其組成主要包含超導磁體、射頻脈衝發射系統、核磁信號接收系統和用於數據採集、儲存、處理以及譜儀控制的計算機系統(如圖)。
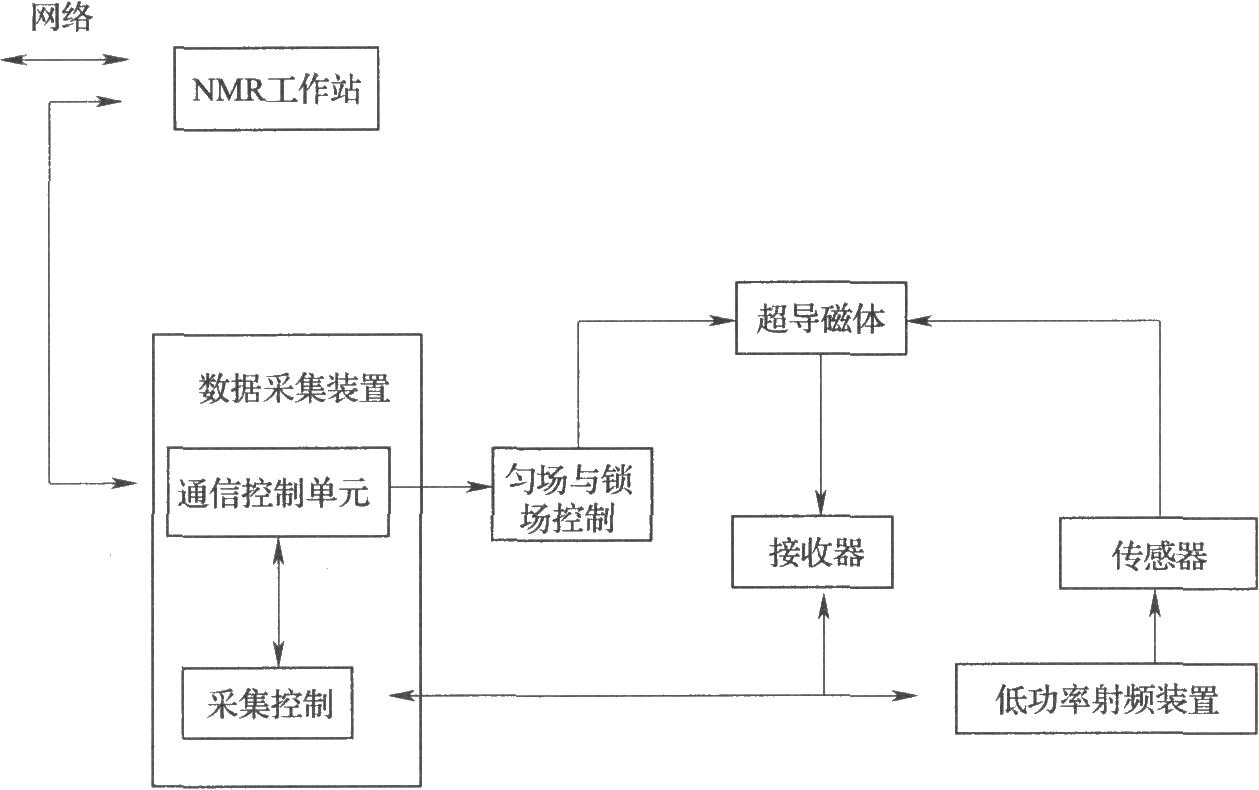
圖PFT核磁共振波譜儀的主要組成
在脈衝核磁共振波譜儀上,一個覆蓋所有共振核的射頻能量的脈衝將同時激發所有的核,當被激發的核回到低能態時產生一個自由感應衰減(FID)信號,它包含所有的時間域信息,經模數轉換後通過計算機進行傅里葉變換得到頻(率)譜。
實驗中按照儀器操作規程設置譜儀參數,如脈衝傾倒角和與之對應的脈衝強度、脈衝間隔時間、數據採樣點(分辨率)、採樣時間等。採集足夠的FIDs,由計算機進行數據轉換,調整相位使盡可能得到純的吸收峯,用參照物校正化學位移值,用輸出設備輸出譜圖。
6 核磁共振譜
核磁共振信號(峯)可提供四個重要參數:化學位移值、譜峯多重性、偶合常數值和譜峯相對強度。處於不同分子環境中的同類原子核具有不同的共振頻率,這是由於作用於特定核的有效磁場由兩部分構成:由儀器提供的特定外磁場以及由核外電子雲環流產生的磁場(後者一般與外磁場的方向相反,這種現象稱爲“屏蔽”)。處於不同化學環境中的原子核,由於屏蔽作用不同而產生的共振條件差異很小,難以精確測定其絕對值,實際操作時採用一參照物作爲基準,精確測定樣品和參照物的共振頻率差。在核磁共振波譜中,一個信號的位置可描述爲它與另一參照物信號的偏離程度,稱爲化學位移。
共振頻率與外磁場強度H0成正比,磁場強度不同,同一化學環境中的核共振頻率不同。爲了解決這個問題,採用位移常數δ來表示化學位移:
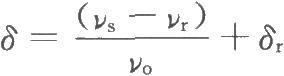
νr爲參照物中磁核的共振頻率;
νo爲儀器的輸出頻率,MHz;
δr爲參照物的化學位移值。
常用的化學位移參照物是四甲基硅烷(TMS),其優點是化學惰性;單峯;信號處在高場,與絕大部分樣品信號之間不會互相重疊干擾;沸點很低(27℃),容易去除,有利於樣品回收。而對於水溶性樣品,常用3-三甲基硅基丙酸鈉-d4(TSP)或2,2-二甲基-2-硅戊基-5-磺酸鈉(DSS),其化學位移值也非常接近於零。DSS的缺點是其三個亞甲基質子有時會干擾被測樣品信號,適於用作外參考。
化學位移僅表示了磁核的電子環境,即核外電子雲對核產生的屏蔽作用,但未涉及同一分子中磁核間的相互作用。這種磁核間的相互作用很小,對化學位移沒有影響,但對譜峯的形狀有着重要影響。這種磁核之間的相互干擾稱爲自旋-自旋偶合,由自旋偶合產生的多重譜峯現象稱爲自旋裂分,裂分間距(赫茲)稱爲偶合常數J,偶合常數與外磁場強度無關。偶合也可發生在氫核與其他核(I≠0)之間,如19F、13C和31P等。
核磁共振信號的另一個特徵是它的強度。在合適的實驗條件下(見“測定方法”),譜峯面積或強度正比於引起此信號的質子數,因此可用於測定同一樣品中不同質子或其他核的相對比例,以及在加入內標後進行核磁共振定量分析。
7 測定方法
在熟悉核磁共振理論的基礎上,應多瞭解樣品的性質,並嚴格遵守操作規程,正確操作儀器。不正確的樣品製備、譜儀調整及參數設置會導致譜圖數據的分辨率和靈敏度降低,甚至給出假峯和錯誤數據。
通常應用最多的是1H(質子)核磁共振波譜,其他還包括19F、31P、13C核磁共振波譜以及各種二維譜等。測定前,一般須先將供試品製成合適的溶液。
7.1 溶劑選擇
合適的溶劑除了對樣品有較好的溶解度外,其殘留的信號峯應不干擾所分析樣品的信號峯。氘代溶劑同時提供異核鎖信號。應儘可能使用高氘代度、高純度的溶劑,並注意氘原子會對其他原子信號產生裂分。常用的核磁共振波譜測定用氘代溶劑及其殘留質子信號的化學位移見下表。
溶劑名稱 | 殘留質子信號δ(ppm) | 可能殘留的水峯δ(ppm)* | |
氘代三氯甲烷 氘代甲醇 氘代丙酮 氘代二甲基亞碸 氘代乙腈 氘代苯 氘代二氧六環 氘代乙酸 氘代三氟乙酸 氘代吡啶 | CDCl3 CD3OD (CD3)2CO DMSO-d6 CD3CN C6D6 D2O 二氧六環-d8 CD3CO2D CF3CO2D C5D5N DMF-d7 | 7.26 3.31 2.05 2.50 1.94 7.16 / 3.55 2.05, 8.5* 12.5* 7.18, 7.55, 8.70 2.77, 2.93, 8.05 | 1.56 4.87 2.84 3.33 2.13 / 4.79 / / / 4.80 / |
*活潑質子的化學位移值是可變的,取決於溫度和溶質的變化。適用於氫譜(1H NMR)的溶劑同樣也適用於氟譜(19FNMR),常見的有CDCl3、CD3OD、D2O、DMSO-d6、DMF-d7、酸和鹼等,通常不含氟的溶劑均可使用。同時應注意含氟樣品中氟原子對其他核的J-偶合。
7.2 樣品製備
按各品種項下的要求。樣品的濃度取決於實驗的要求及儀器的類型,測定非主要成分時需要更高的濃度。供試液的體積取決於樣品管的大小及儀器的要求,通常樣品溶液的高度應達到線圈高度的2倍以上。選用符合定量要求的核磁管,常用外徑爲5mm或10mm,長度爲15cm或20cm的核磁管。當樣品量較少時可選用微量核磁管。
7.3 測定
將樣品管放入譜儀中,先進行樣品和譜儀的調諧,再仔細對譜儀勻場,使譜儀達到最佳工作狀態。設置合適的實驗參數,採樣,完成後再進行圖譜處理,並分段積分。
同一個實驗通常可同時得到定性和定量數據。對於核磁共振定量分析,實驗參數的正確設置非常重要,以保證每個峯的積分面積與質子數成正比。必須保證有足夠長的弛豫時間,以使所有激發核都能完全弛豫,因而定量分析通常需要更長的實驗時間。
7.4 定性和定量分析
核磁共振波譜分析可廣泛應用於結構確證,熱力學、動力學和反應機理的研究,以及用於定量分析。
7.4.1 1.定性分析
核磁共振波譜是一個非常有用的結構解析工具,化學位移提供原子核環境信息,譜峯多重性提供相鄰基團情況以及立體化學信息,偶合常數值大小可用於確定基團的取代情況,譜峯強度(或積分面積)可確定基團中質子的個數等。一些特定技術,如雙共振實驗、化學交換、使用位移試劑、各種二維譜等,可用於簡化複雜圖譜、確定特徵基團以及確定偶合關係等。
對於結構簡單的樣品可直接通過氫譜的化學位移值、偶合情況(偶合裂分的峯數及偶合常數)及每組信號的質子數來確定,或通過與文獻值(圖譜)比較確定樣品的結構,以及是否存在雜質等。與文獻值(圖譜)比較時,需要注意一些重要的實驗條件,如溶劑種類、樣品濃度、化學位移參照物、測定溫度等的影響。對於結構複雜或結構未知的樣品,通常需要結合其他分析手段,如質譜等方能確定其結構。
7.4.2 2.定量分析
與其他核相比,1H核磁共振波譜更適用於定量分析。在合適的實驗條件下,兩個信號的積分面積(或強度)正比於產生這些信號的質子數:

式中A1、A2爲相應信號的積分面積(或強度);N1、N2爲相應信號的總質子數。
如果兩個信號來源於同一分子中不同的官能團,式(1)可簡化爲:

式中,n1、n2分別爲相應官能團中的質子數。如果兩個信號來源於不同的化合物,則
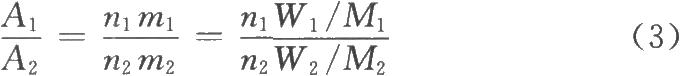
式中m1、m2分別爲化合物1和化合物2的分子個數;W1、W2分別爲其質量;
M1、M2分別爲其分子量。
由式(2)和(3)可知,核磁共振波譜定量分析可採用絕對定量和相對定量兩種模式。
在絕對定量模式下,將已精密稱定重量的樣品和內標混合配製溶液,測定,通過比較樣品特徵峯的峯面積與內標峯的峯面積計算樣品的含量(純度)。合適的內標應滿足如下要求:有合適的特徵參考峯,最好是適宜寬度的單峯;內標物的特徵參考峯與樣品峯分離;能溶於分析溶劑中;其質子是等權重的;內標物的分子量與特徵參考峯質子數之比合理;不與待測樣品相互作用等。常用的內標物有:1,2,4,5-四氯苯、1,4-二硝基苯、對苯二酚、對苯二酸、苯甲酸苄酯、順丁烯二酸等。內標物的選擇依據樣品性質而定。
相對定量模式主要用於測定樣品中雜質的相對含量(或混合物中各成分相對含量),由式(3)來計算。
7.4.2.1 (1)絕對定量模式
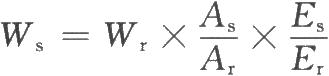
式中Wr爲內標物的重量;
As和Ar分別爲供試品特徵峯和內標峯的平均峯面積;
Es和Er分別爲供試品和內標物的質子當量重量(質量)(以分子量除以特徵峯的質子數計算得到)。
7.4.2.2 (2)相對定量模式
由下式計算供試品中各組分的摩爾百分比:式中A1和A2分別爲各品種項下所規定的各特徵基團共振峯的平均峯面積;n1、n2分別爲各特徵基團的質子數。
8 參考資料
- ^ [1] 國家藥典委員會.中華人民共和國藥典:2010年版:二部[M].北京:中國醫藥科技出版社,2010.