1 概述
範科尼綜合徵(Fanconi syndrome)也稱Fanconi-de Toni綜合徵、骨軟化-腎性糖尿-氨基酸尿-高磷酸尿綜合徵、多種腎小管功能障礙性疾病。是指遺傳性或獲得性近端腎小管的功能異常引起的一組徵候羣。臨牀表現爲腎性過多丟失而產生的全氨基酸尿、磷酸鹽尿、葡萄糖尿、碳酸氫鹽尿以及尿酸等有機酸尿;過多丟失電解質而產生的各種代謝性併發症,如低磷血癥、低鈣血癥、高氯性代謝性酸中毒、維生素D缺乏病、骨質疏鬆、脫水、生長遲緩等;因丟失分子量小於5萬的蛋白質而產生腎小管性蛋白尿。通過對氨馬尿酸清除試驗顯示腎小管排泄功能障礙,腎小球功能基本正常或與酸中毒相比不成比例。近年有人將抗利尿激素抵抗的多尿症也包括在其中,但以氨基酸尿、糖尿、磷酸鹽尿爲基本診斷指標。
範科尼綜合徵國內罕見,嬰兒與成人均可發病,起病緩慢,多於青壯年期出現症狀,有腎性糖尿、多種氨基酸尿、高鈣尿症、腎丟失鈉、低磷血癥、近端腎小管性酸中毒、低尿酸血癥、腎小管性蛋白尿,低鉀血癥(肌無力、軟癱、週期性癱瘓等),低鈣血癥(手足搐搦症)等。常見併發症爲腎小管性酸中毒;低鉀血癥;繼發性甲狀旁腺功能亢進、腎性骨病、骨畸形、骨軟化症;腎結石等。本綜合徵病因複雜,預後與根底病息息相關,如及時採取適當治療,一般預後尚好。但有些根底病無法根治,故預後不良,常因繼發感染或腎功能衰竭而於兒童期死亡。
8 病因
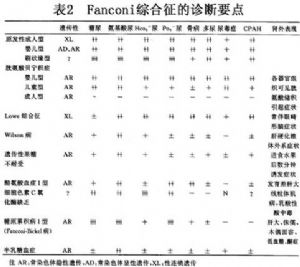
範科尼綜合徵的病因很多,可分爲原發性與繼發性兩類。原發性Fanconi綜合徵又分爲:嬰兒型、成人型以及刷狀緣缺失型三種類型。繼發性Fanconi綜合徵又包括繼發於遺傳性疾病與繼發於後天獲得性疾病。前者包括:胱氨酸儲積病、酪氨酸血癥Ⅰ型、糖原貯積病ⅠⅠ型、半乳糖血症、遺傳性果糖不耐受、細胞色素C氧化酶缺乏症、Wilson病、Lowe綜合徵、遺傳性成骨不全、Alport綜合徵、先天性腎病綜合徵、維生素D依賴性佝僂依賴性佝僂病等;後者包括:腎病綜合徵、移植腎、急慢性間質性腎炎、多發性骨髓瘤腎病、舍格倫綜合徵、腎澱粉樣變性、重金屬中毒、藥物(過期四環素、氨基糖類抗生素、6-巰基嘌呤、順鉑等)引起的腎損害、低鉀性腎病、甲狀旁腺功能亢進以及腫瘤相關性腎病等。病因詳見表1。

9 發病機制
Fanconi綜合徵發病機制尚未完全闡明。目前認爲不同於單項物質轉運異常,即不是由於某種特異性的載體或受體缺陷所致。主要有兩方面機制:
9.1 腎小管細胞膜有漏隙,不能使溶質充分再吸收
反漏的證據是腎性糖尿屬A型,表明葡萄糖轉運再吸收部位較少,磷酸鹽、碳酸氫鹽在濾過負荷減少的情況下仍有丟失。這表明它們的排泌是通過腎小管的泄漏。
9.2 腎小管內能量代謝不足,產生的能量難以支持正常轉運
有些毒物或藥物中毒以及遺傳代謝疾病使某些代謝產物在腎小管內儲積過多影響了細胞內氧化磷酸化過程,ATP生成不足,沒有足夠的能量支持腎小管轉運物質。無論什麼機制可最終導致多種物質轉運異常。範可尼綜合徵是近曲小管多項轉運缺陷病,包括氨基酸、葡萄糖、鈉、鉀、鈣、磷、碳酸氫鈉、尿酸和蛋白質。原發性者近端小管呈天鵝頸樣變形。
10 範科尼綜合徵的臨牀表現
範科尼綜合徵較罕見,多於成年出現症狀,有腎性糖尿、多種氨基酸尿、高鈣尿症、腎丟失鈉、低磷血癥、近端腎小管性酸中毒、低尿酸血癥、腎小管性蛋白尿,低鉀血癥(肌無力、軟癱、週期性癱瘓等),低鈣血癥(手足搐搦症)等。長期低鈣血癥,可引起繼發性甲狀旁腺功能亢進、腎性骨病。本病最突出的臨牀表現爲小兒維生素D缺乏缺乏病和成人的骨軟化症。繼發性範可尼綜合徵的臨牀表現,基本上與原發性者同,但可有其根底疾病的臨牀表現。本綜合徵臨牀表現複雜,根據其臨牀類型分述如下:
10.1 原發性Fanconi綜合徵
原發性Fanconi綜合徵包括3種類型:
10.1.1 (1)成人型Fanconi綜合徵
10~20歲以後起病,有多種腎小管功能障礙,如全氨基酸尿、糖尿、磷酸鹽尿、高血氯性酸中毒、低鉀血癥等。突出的症狀是軟骨病,少數病例可有酮症晚期可出現腎功能衰竭。
10.1.2 (2)嬰兒型Fanconi綜合徵
多於6~12個月發病,多尿、煩渴、脫水、便祕、無力、拒食、發熱,生長發育遲緩腎性氨基酸尿,可有抗維生素D佝僂佝僂病及嚴重營養不良現象。實驗室檢查呈低血鉀、低血磷、低血鈣及鹼性磷酸酶增高、高氯性代謝性酸中毒、尿中可滴定酸及NH4可減少,尿糖微量或4~5g/d,血糖正常,急性起病者預後差,常死於尿毒症。慢性起病者多於2歲以後發病,症狀較輕,突出表現爲侏儒和(或)抗維生素D佝僂佝僂病。
10.1.3 (3)特發性刷狀緣缺失型Fanconi綜合徵
1984年Manz等首次報道1例小兒由於近曲小管刷狀緣完全缺失而引起Fanconi綜合徵,因爲葡萄糖及各種氨基酸載運系統完全喪失,故這些物質的清除率近於腎小球濾過率。
10.2 繼發性Fanconi綜合徵
繼發性Fanconi綜合徵多有原發病,不同病因引起者表現各有不同。
10.2.1 (1)胱氨酸儲積症
胱氨酸儲積症又稱Lignac-Fanconi綜合徵,系胱氨酸沉着於細胞溶酶體而表現爲Fanconi綜合徵。正常人細胞內溶酶體是細胞內蛋白降解的部位,細胞內蛋白降解產生氨基酸通過溶酶體膜轉輸系統輸入胞質而被再利用。本病因溶酶體內胱氨酸運載體有缺陷,使胱氨酸在溶酶體中儲積,從而破壞了溶酶體的完整性,並可使具有破壞性的溶酶體酶漏至細胞漿,影響了功能。本病與胱氨酸尿症不同,後者是腎小管上皮轉運胱氨酸障礙,只引起胱氨酸尿,前者則引起許多器官細胞內胱氨酸儲積,腎臟是主要受累器官之一。
胱氨酸儲積病所引起的Fanconi綜合徵不同於其他原因所致Fanconi綜合徵,常以失鉀、脫水、多飲、滲透性利尿爲突出表現。臨牀上可分3型:
①嬰兒型或腎病型:胱氨酸沉積於各種組織溶酶體內,在白細胞內可能比正常大80倍,腎臟髓質可能沉着近100倍,因腎小管損傷出現各種症狀,患兒多於6個月左右開始發病,多尿、煩渴、便祕、多飲、嘔吐、拒食、消瘦、發育障礙,由於脫水有反覆發熱,可發生維生素D缺乏缺乏病及侏儒症。由於角膜、結膜的胱氨酸沉着而畏光,眼底周圍色素脫失,可引起末梢視網膜病變。此外尚可表現爲甲狀腺功能低下、糖尿病、脾大、腦水腫、肌病等。腎小管功能障礙表現爲腎濃縮功能障礙、氫離子排泄功能障礙,而至尿液不能酸化至pH 5.5以下,呈腎小管性酸中毒。
②兒童型或中間型:10歲左右發病,進展較慢,骨病不嚴重,無侏儒症。組織胱氨酸含量遠較嬰兒型爲低,白細胞內胱氨酸含量爲正常的30倍,也可表現爲腎臟病變,甚至發展爲尿毒症,骨骼畸形、畏光、視網膜病變、胱氨酸引起的脾大也可產生。Fanconi綜合徵表現不明顯。
③成人型:無腎病表現,以其他器官功能障礙爲主。成人型又可分爲急性與慢性,前者與嬰兒型類似,後者與兒童型類似。
通過骨髓片、白細胞、直腸黏膜中的結晶分析或裂隙燈檢查角膜有胱氨酸結晶而診斷。嬰兒型因患兒拒食引起飢餓性酮症加上腎小管性糖尿,易誤診爲幼兒糖尿病,需提高警惕。
10.2.2 (2)Lowe綜合徵
Lowe綜合徵系Lowe 1952年首先報道,亦稱眼-腦-腎綜合徵,臨牀表現特點爲:
①眼症狀:先天性白內障(雙側)伴有先天性青光眼(牛眼)、視力嚴重障礙、眼球震顫及畏光。
②腦症狀:嚴重智力發育遲緩,肌張力低、腱反射減弱或消失,患兒常哭泣樣尖叫。
③腎小管功能障礙:多組氨基酸尿、磷酸鹽尿、碳酸氫鹽尿、尿酸化功能差,尿中排出賴氨酸、酪氨酸爲多。還可有腎小管性蛋白尿、後期可發生慢性腎功能不全。按自然發展可分爲3期:嬰兒期,以眼腦症狀爲主。表現爲頭顱畸形(長頭,前額高出,鞍鼻,高齶弓等);兒童期,出現不完全Fanconi綜合徵,有腎小管性蛋白尿,嚴重磷酸鹽尿可引起抗維生素D佝僂佝僂病或骨質疏鬆。一般情況下有較輕或無糖尿、失鉀及多尿。常有臍疝、隱睾畸形,以及特殊的手指小關節炎;成人期,腎小管病症狀消退,出現腎功能不全或營養不良,常合併肺炎而死亡。本綜合徵主要是對症治療,如糾正腎小管性酸中小管性酸中毒,抗維生素D佝僂佝僂病的治療等。無根治辦法,預後不良。常因繼發感染或腎功能衰竭而於兒童期死亡。
肝豆狀核變性(Wilson病)系少見的隱性遺傳性代謝性疾病。因爲血漿銅藍蛋銅藍蛋白(ceruloplasmin)含量降低,銅氧化酶活性降低導致腸道大量吸收銅,大量銅沉積於肝、腦、角膜、腎小管引起相應的症狀。銅沉積於腦及肝引起錐體外系神經症狀及肝硬化,銅沉積於角膜引起Kayser-Fleischer環。銅沉積於近端腎小管及遠端腎小管引起Fanconi綜合徵,可伴有重碳酸鹽丟失和腎鈣質沉着伴高鈣尿症,腎小管性酸中毒,腎鈣化,腎結石。
範科尼綜合徵可用青黴胺治療,促進銅從尿中排出但停用後會復發。其他治療如二巰丙醇(BAL)可增加銅排出,口服硫化鈉可改善神經系統和肝症狀,但腎小管病變無改善,骨化醇可治療骨病變。
10.2.3 (4)遺傳性果糖不耐受
遺傳性果糖不耐受爲常染色體隱性遺傳的酶缺乏病。因肝、腎組織中缺乏1-磷酸果糖醛縮酶或者1,6-二磷酸果糖醛縮酶的活性下降,從而使1-磷酸果糖不能裂解而儲積於細胞內產生病變,同時因不能產生ATP而影響細胞的能量代謝。若給患者輸注果糖可產生Fanconi綜合徵複合腎小管功能障礙,若杜絕果糖則腎小管功能正常。本病的發病機制可能是由於腎皮質細胞內降解1-磷酸果糖的醛縮酶缺乏,小管上皮細胞內磷酸鹽減少,對腺苷脫氨酶(ADA)的抑制作用減弱,以至ADA活性增強,使腺苷脫氨生成次黃苷(inosine),經核苷磷酸化酶作用生成次黃嘌呤,再由黃嘌呤氧化酶作用生成尿酸。故產生低磷高尿酸血癥。另外,由於磷可增加腎皮質中ATP產生率,使果糖轉化爲α-磷酸甘油,當低磷血癥時,ATP生成減少,也會影響到能量供應。由此可見1-磷酸果糖不是毒性代謝產物,它不抑制酶系統,而是由於磷的耗空使ATP及其他高能磷酸化合物在腎小管細胞某些位點的產生受到很大限制。
嬰兒期因攝食乳糖無症狀,當食用果糖或水果時急性發病,攝食後20~40min出現嘔吐、腹瀉、低血糖與高尿酸血癥,2h後出現急性Fanconi綜合徵,乳酸性酸中毒、高膽紅素血癥、肝大。及時停止攝入果糖,治療低血糖,病情可能緩慢逆轉,否則可直接威脅生命。
10.2.4 (5)酪氨酸血癥(tyrosinemia)
酪氨酸血癥(tyrosinemia)是由於患者缺少對羥苯丙酮酸氧化酶(hydroxylphenyl puruvic acid oxidase)而導致酪氨酸代謝異常可引起Fanconi綜合徵。其特徵是血中酪氨酸、苯丙氨酸、蛋氨酸、丙氨酸顯著增加,其他氨基酸很少增加。在尿中排出酪氨酸、苯丙氨酸、蛋氨酸和對羥苯丙酮酸,對羥苯乙酸的酚酸代謝產物也增加。臨牀上本病分成兩型:Ⅰ型酪氨酸血癥即爲暫時性高酪氨酸血癥。若投以酪氨酸會發生肝腎功能複合損害,長期持續則腎皮質腎小管發生變性,肝硬化伴門脈高壓和腹水。有的病例出現維生素D缺乏缺乏病,白內障形成或由於胰島細胞肥大而引起低血糖症。Ⅱ型的特徵爲持續性高酪氨酸血癥,病情持續發展,有嚴重智力障礙,皮膚異常、白內障、生長緩慢,而無明顯的肝腎損害,其對羥苯丙酮酸氧化酶活性正常。
飲食治療(如低酪氨酸、低苯丙氨酸飲食)可改善Ⅱ型患者病情,對Ⅰ型患者可減輕腎小管損害,但對嚴重肝損害無效。
10.2.5 (6)細胞色素C氧化酶缺乏症
細胞色素C氧化酶缺乏症可引起Fanconi綜合徵,這是因爲腎小管上皮細胞線粒體中缺乏該酶而使電子傳遞鏈中ATP合成及氧化磷酸化過程障礙。患者多在出生後11~13周發病,主要表現爲線粒體肌病,乳酸性酸中毒及腎性糖尿、氨基酸尿、磷酸鹽尿等腎小管功能障礙。
10.2.6 (7)多發性骨髓瘤所致Fanconi綜合徵
多發性骨髓瘤可伴有腎澱粉樣變性或輕鏈蛋白(κ或λ)引起腎小管損傷而致非遺傳性繼發性Fanconi綜合徵。臨牀特徵爲骨痛、肌無力、疲乏、貧血、骨軟化症、假性骨折等,並有葡萄糖尿氨基酸尿、磷酸鹽尿、腎性尿崩症、腎小管性酸中毒等腎小管功能不全的表現。其中Fanconi綜合徵爲多發性骨髓瘤的伴發症狀。
10.2.7 (8)毒性物質引起的Fanconi綜合徵
毒性物質可引起繼發性Fanconi綜合徵。例如過期的四環素其降解產物具有腎小管毒性。其臨牀特徵爲肌病、眩暈、酸中毒、多尿、低鉀血癥。雖然停藥後可能恢復,但有的病程可持續2年以上。
15 鑑別診斷
1.嬰兒型Fanconi綜合徵鑑別診斷應注意其他原因所致的腎小管性酸中毒,肌無力症狀或步態不穩類似神經系統病變或原發性肌病。也極似Fanconi綜合徵的嬰兒型應注意區分。
2.成人因其他代謝性骨病引起骨質疏鬆伴肌病也類似Fanconi綜合徵;尿毒症患者可有葡萄糖尿或氨基酸尿而無低磷酸鹽血癥;Wilson病也會與運動系疾病相混淆。總之,複合性腎小管排出溶質過多必須尋找其原發疾病。
16 範科尼綜合徵的治療
16.1 病因治療
繼發性Fanconi綜合徵應治療基礎疾病。Wilson病或重金屬中毒可通過促進毒物排泄,遺傳代謝病通過飲食管理減少代謝毒性物質沉積,減輕對腎小管的損害。胱氨酸儲積症繼發性Fanconi綜合徵,應給予低胱氨酸飲食及對症治療。骨病變可用維生素D225萬~50萬U或維生素D332000~1000U或羥膽骨化醇200~5000U。有脫水及酸中毒應作相應處理。早期可用枸櫞酸鉀鈉溶液10~15ml口服,3~5次/d。青黴胺可試用於消除胱氨酸,但不能減少細胞內胱氨酸沉着;Dithiothreitol(DDT)療效欠佳,半胱氨酸效果較好。
16.2 對症治療
(1)糾正酸中毒:根據碳酸氫根丟失情況補充鹼劑,2~10mmol/(kg·d),可用重碳酸氫鹽、枸櫞酸鹽、乳酸鹽等,4~5次/d,分次給服,以血中碳酸氫鹽水平恢復正常爲標準。補鈉可使低血鉀加重,應注意檢測;對已有低血鉀者宜同時補鉀2~4mmol/(kg·d)。若鹼劑用量過大患者不能耐受時,可加用氫氯噻嗪(雙氫克尿塞)2~3mg/(kg·d),它可使細胞外液縮減而促進碳酸氫根的再吸收,但應謹慎防止腎小球濾過率下降。
(2)糾正低血容量:Fanconi綜合徵常因多尿而致脫水,除了針對病因治療外,應補足含鹽的液體(包括鈉、鉀、鈣等),可採用定時口服,必要時臨時追加。
(3)糾正低磷血癥:應用中性磷酸鹽1~3g/d,分5次服用。如有腹瀉或腹部不適可減量。注意補磷可加重低血鈣與骨病,故應合用維生素D 5000U/d或1,25(OH)2D30.25~0.5μg/d,應從少量開始,逐漸加至足量。爲防止腎鈣化應監測尿鈣排量,以不超過0.6g/d爲標準。